Section 3 Candidate prioritization
3.1 Jackknife-based validation of PURF models
3.1.1 Analysis
In Python:
import pandas as pd
import numpy as np
import pickle
from sklearn.impute import SimpleImputer
from purf.pu_ensemble import PURandomForestClassifier
from sklearn.preprocessing import MinMaxScaler
from scipy.spatial import distance
from joblib import Parallel, delayed
from sklearn.ensemble._forest import _generate_unsampled_indices
import os
import re
import session_info# input set (5393, 1 + 272)
data = pd.read_csv('./data/supplementary_data_3_pf_ml_input.csv', index_col=0)
pf3d7_features = data.iloc[:,1:]
pf3d7_outcome = np.array(data.antigen_label)
REF_ANTIGENS = {'CSP': 'PF3D7_0304600.1-p1', 'RH5': 'PF3D7_0424100.1-p1', 'MSP5': 'PF3D7_0206900.1-p1', 'P230': 'PF3D7_0209000.1-p1'}# private function for train_purf()
def _get_ref_antigen_stats(idx, tree, X, y, ref_indices, max_samples=None):
if max_samples is None:
max_samples = y.shape[0]
oob_indices = _generate_unsampled_indices(tree.random_state, y.shape[0], max_samples)
ref_oob = [i in oob_indices for i in ref_indices]
ref_pred = list()
pred = tree.predict_proba(X[ref_indices,:], check_input=False)
ref_pred = pred[:,1]
return ref_oob, ref_preddef train_purf(features, outcome, res_path, pickle_path, tree_filtering=None, model_path=None, n_jobs=1):
# Imputation
imputer = SimpleImputer(strategy='median')
X = imputer.fit_transform(features)
X = pd.DataFrame(X, index=features.index, columns=features.columns)
y = outcome
features = X
print('There are %d positives out of %d samples before feature space weighting.' % (sum(y), len(y)))
# Feature space weighting
lab_pos = X.loc[y==1,:]
median = np.median(lab_pos, axis=0)
# Feature space weighting
lab_pos = X.loc[y==1,:]
median = np.median(lab_pos, axis=0)
scaler = MinMaxScaler(feature_range=(1,10))
dist = list()
for i in range(lab_pos.shape[0]):
dist.append(distance.euclidean(lab_pos.iloc[i, :], median))
dist = np.asarray(dist).reshape(-1, 1)
counts = np.round(scaler.fit_transform(dist))
counts = np.array(counts, dtype=np.int64)[:, 0]
X_temp = X.iloc[y==1, :]
X = X.iloc[y==0, :]
y = np.asarray([0] * X.shape[0] + [1] * (sum(counts)))
appended_data = [X]
for i in range(len(counts)):
appended_data.append(pd.concat([X_temp.iloc[[i]]] * counts[i]))
X = pd.concat(appended_data)
print('There are %d positives out of %d samples after feature space weighting.' % (sum(y), len(y)))
res = pd.DataFrame({'protein_id': X.index, 'antigen_label' : y})
if tree_filtering is not None:
# get ref antigen indices
ref_index_dict = {ref:list() for ref in list(REF_ANTIGENS.values())}
for i in range(res.shape[0]):
if res['protein_id'][i] in list(REF_ANTIGENS.values()):
ref_index_dict[res['protein_id'][i]].append(res.index[i])
ref_indices = sum(ref_index_dict.values(), [])
# get OOB stats and predictions
X = X.astype('float32')
purf = pickle.load(open(model_path, 'rb'))
trees = purf.estimators_
idx_list = [i for i in range(len(trees))]
stats_res = Parallel(n_jobs=n_jobs)(
delayed(_get_ref_antigen_stats)(idx, trees[idx], np.array(X), y, ref_indices) for idx in idx_list)
# ref_oob data structure:
# rows represent individual trees
# column represent reference antigens
# cells indicate whether the reference antigen is in the OOB samples of the tree
ref_oob = np.array([ref_oob for ref_oob, ref_pred in stats_res])
# ref_pred data structure:
# rows represent individual trees
# column represent reference antigens
# cells indicate the prediction of the reference antigen by the tree
ref_pred = np.array([ref_pred for ref_oob, ref_pred in stats_res])
# analyze duplicated reference antigens as a group
cumsum_num_ref = np.cumsum(np.array([len(v) for k,v in ref_index_dict.items()]))
ref_oob_all = np.array([ref_oob[:, 0:cumsum_num_ref[i]].any(axis=1) if i == 0 else \
ref_oob[:, cumsum_num_ref[i - 1]:cumsum_num_ref[i]].any(axis=1) \
for i in range(len(REF_ANTIGENS))]).T
ref_pred_all = np.array([ref_pred[:, 0:cumsum_num_ref[i]].any(axis=1) if i == 0 else \
ref_pred[:, cumsum_num_ref[i - 1]:cumsum_num_ref[i]].sum(axis=1) \
for i in range(len(REF_ANTIGENS))]).T
# calculate number of reference antigens as OOB samples for each tree
oob_total = ref_oob_all.sum(axis=1)
# assign score of 1 to trees that correctly predict all OOB reference antigens; otherwise, assign 0 score
weights = np.zeros(len(trees))
# iterate through the trees and calculate the stats
for i in range(len(trees)):
oob_list = list(ref_oob_all[i,:])
pred_list = list(ref_pred_all[i,:])
if oob_total[i] == 0:
weights[i] = 0
else:
if sum(np.array(pred_list)[oob_list] != 0) == oob_total[i]:
weights[i] = 1
if tree_filtering is None:
# Training PURF
purf = PURandomForestClassifier(
n_estimators = 100000,
oob_score = True,
n_jobs = 64,
random_state = 42,
pos_level = 0.5
)
purf.fit(X, y)
else:
purf._set_oob_score_with_weights(np.array(X), y.reshape(-1,1), weights=weights)
# Storing results
res['OOB score'] = purf.oob_decision_function_[:,1]
features = features.merge(res.groupby('protein_id').mean(), left_index=True, right_on='protein_id')
features = features[['antigen_label', 'OOB score']]
features.to_csv(res_path)
if tree_filtering is None:
with open(pickle_path, 'wb') as out:
pickle.dump(purf, out, pickle.HIGHEST_PROTOCOL)
else:
with open(pickle_path, 'wb') as out:
pickle.dump({'model': purf, 'weights': weights}, out, pickle.HIGHEST_PROTOCOL)3.1.1.1 Training on whole data set
In Python:
train_purf(pf3d7_features, pf3d7_outcome,
res_path='~/Downloads/pos_level/0.5_res_tree_filtering.csv',
pickle_path='~/Downloads/pos_level/0.5_purf_tree_filtering.pkl',
model_path='~/Downloads/pos_lebel/0.5_purf.pkl',
n_jobs=1)
wo_tree_filtering = pd.read_csv('~/Downloads/pos_level/0.5_res.csv', index_col=0)
w_tree_filtering = pd.read_csv('~/Downloads/pos_level/0.5_res_tree_filtering.csv', index_col=0)
merged_df = pd.concat([wo_tree_filtering, w_tree_filtering['OOB score']], join='outer', axis=1)
merged_df.columns = ['antigen_label', 'oob_score_without_tree_filtering', 'oob_score_with_tree_filtering']
merged_df.to_csv('./data/supplementary_data_4_purf_oob_predictions.csv')3.1.1.2 Validation for PURF without tree filtering
In Python:
for (idx, (antigen, out)) in enumerate(zip(pf3d7_features.index, pf3d7_outcome)):
if out == 1:
if antigen in REF_ANTIGENS.values():
continue
pf3d7_outcome_ = pf3d7_outcome.copy()
pf3d7_outcome_[idx] = 0
train_purf(pf3d7_features, pf3d7_outcome_,
res_path='~/Downloads/jackknife/' + antigen + '_res.csv',
pickle_path='~/Downloads/jackknife/' + antigen + '_purf.pkl')dir = '~/Downloads/jackknife/'
files = os.listdir(dir)
for file in files:
if file.endswith('csv'):
tmp = pd.read_csv(dir + file, index_col=0)['antigen_label']
break
data_frames = [pd.read_csv(dir + file, index_col=0)['OOB score'] for file in files if file.endswith('csv')]
merged_df = pd.concat([tmp] + data_frames, join='outer', axis=1)
colnames = ['antigen_label'] + [re.match('PF3D7_[0-9]+\.[0-9]-p1', file)[0] for file in files if file.endswith('csv')]
merged_df.columns = colnames
merged_df.to_csv('./data/supplementary_data_5_validation.csv')3.1.1.3 Validation for PURF with tree filtering
In Python:
for (idx, (antigen, out)) in enumerate(zip(pf3d7_features.index, pf3d7_outcome)):
if out == 1:
if antigen in REF_ANTIGENS.values():
continue
pf3d7_outcome_ = pf3d7_outcome.copy()
pf3d7_outcome_[idx] = 0
train_purf(pf3d7_features, pf3d7_outcome_,
res_path='~/Downloads/tree_filtering_jackknife/' + antigen + '_res.csv',
pickle_path='~/Downloads/tree_filtering_jackknife/' + antigen + '_purf.pkl',
model_path='~/Downloads/jackknife/' + antigen + '_purf.pkl',
n_jobs=1)dir = '~/Downloads/tree_filtering_jackknife/'
files = os.listdir(dir)
for file in files:
if file.endswith('csv'):
tmp = pd.read_csv(dir + file, index_col=0)['antigen_label']
break
data_frames = [pd.read_csv(dir + file, index_col=0)['OOB score'] for file in files if file.endswith('csv')]
merged_df = pd.concat([tmp] + data_frames, join='outer', axis=1)
colnames = ['antigen_label'] + [re.match('PF3D7_[0-9]+\.[0-9]-p1', file)[0] for file in files if file.endswith('csv')]
merged_df.columns = colnames
merged_df.to_csv('./data/supplementary_data_6_tree_filtering_validation.csv')3.1.2 Plotting
In R:
library(mixR)
library(pracma)
library(rlist)
library(ggplot2)
library(cowplot)
library(grid)
library(ggbeeswarm)
library(ggpubr)data <- read.csv("./data/supplementary_data_4_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)
# Extract data with only unlabeled proteins
data_unl <- data[data$antigen_label == 0, ]
validation_wo_tree_filtering <- read.csv("./data/supplementary_data_5_validation.csv", row.names = 1, check.names = FALSE)
validation_w_tree_filtering <- read.csv("./data/supplementary_data_6_tree_filtering_validation.csv", row.names = 1, check.names = FALSE)3.1.2.1 Plot score distribution
fit <- mixfit(data_unl[, "oob_score_with_tree_filtering"], ncomp = 2)
# Calculate receiver operating characteristic (ROC) curve
# for putative positive and negative samples
x <- seq(-0.5, 1.5, by = 0.01)
neg_cum <- pnorm(x, mean = fit$mu[1], sd = fit$sd[1])
pos_cum <- pnorm(x, mean = fit$mu[2], sd = fit$sd[2])
fpr <- (1 - neg_cum) / ((1 - neg_cum) + neg_cum) # false positive / (false positive + true negative)
tpr <- (1 - pos_cum) / ((1 - pos_cum) + pos_cum) # true positive / (true positive + false negative)
p1 <- plot(fit, title = paste0("PURF with tree filtering", " (AUROC = ", round(trapz(-fpr, tpr), 2), ")")) +
scale_fill_manual(values = c("blue", "red"), labels = c("Putative negative", "Putative positive")) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = c(0.8, 0.9),
legend.background = element_blank()
) +
xlim(-0.3, 1.3) +
xlab("Score (proportion of votes)") +
ylab("Density")3.1.2.2 Percent rank for labeled positives
data_ <- data[c("antigen_label", "oob_score_with_tree_filtering")]
colnames(data_) <- c("antigen_label", "OOB score")
data_$percent_rank <- rank(data_[["OOB score"]]) / nrow(data)
data_ <- data_[data$antigen_label == 1, ]
data_ <- data_[order(-data_$percent_rank), ]
data_$x <- 1:nrow(data_) / nrow(data_)
cat(paste0("EPR: ", sum(data_$`OOB score` >= 0.5) / nrow(data_), "\n"))
p2 <- ggplot(data_, aes(x = x, y = `percent_rank`)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "black") +
geom_line() +
geom_point(aes(color = `OOB score`), size = 1) +
scale_colour_gradient2(low = "blue", mid = "purple", high = "red", midpoint = 0.5, limits = c(0, 1)) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.35, 0.15),
legend.background = element_blank()
) +
guides(colour = guide_colourbar(title.position = "top", direction = "horizontal")) +
ggtitle(paste0("PURF with tree filtering", " (AUC = ", round(trapz(c(0, data_$x, 1), c(1, data_$percent_rank, 0)), 2), ")")) +
ylim(0, 1) +
xlab("Ranked known antigens (scaled)") +
ylab("Percent rank") +
labs(colour = "Score (proportion of votes)")3.1.2.3 Comparison of known antigens
calculate_known_antigen_scores <- function(validation_data, baseline_scores) {
scores <- c()
for (i in 2:ncol(validation_data)) {
known_antigen <- sort(colnames(validation_data))[i]
other_antigens <- sort(colnames(validation_data))[-c(1, i)]
scores <- c(scores, mean(validation_data[other_antigens, known_antigen] - baseline_scores[other_antigens, ]))
}
return(scores)
}
scores_wo_tree_filtering <- calculate_known_antigen_scores(
validation_wo_tree_filtering,
data["oob_score_without_tree_filtering"]
)
scores_w_tree_filtering <- calculate_known_antigen_scores(
validation_w_tree_filtering,
data["oob_score_with_tree_filtering"]
)
data_ <- data.frame(
group = factor(c(
rep(0, length(scores_wo_tree_filtering)),
rep(1, length(scores_w_tree_filtering))
)),
score = c(scores_wo_tree_filtering, scores_w_tree_filtering),
paired = rep(1:48, 2)
)
stats <- compare_means(score ~ group, data = data_, method = "wilcox.test", paired = TRUE)
p3 <- ggplot(data_, aes(x = group, y = score)) +
geom_hline(yintercept = 0, color = "black", linetype = "dashed") +
geom_boxplot(aes(color = group), outlier.color = NA, lwd = 1.5, show.legend = FALSE) +
geom_line(aes(group = paired), alpha = 0.6, color = "grey80") +
geom_beeswarm(aes(fill = group),
color = "black", alpha = 0.5, size = 2, cex = 2, priority = "random",
shape = 21
) +
scale_color_manual(values = c("#f7d59e", "#fcd7d7")) +
scale_fill_manual(values = c("#fc9d03", "red"), labels = c("Without tree filtering", "With tree filtering")) +
scale_x_discrete(labels = c("Without tree filtering", "With tree filtering")) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(10, 5, 18, 5, "pt"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = "none"
) +
ylab("Mean difference in scores (proportion of votes)")3.1.2.4 Comparison of top 200 candidates
calculate_overlapping_candidates <- function(data, validation_data) {
top_200 <- list()
for (i in 1:48) {
data_ <- validation_data[data$antigen_label == 0, ]
data_ <- data_[order(-data_[, 1 + i]), ][1:200, ]
top_200 <- list.append(top_200, rownames(data_))
}
union_top_200 <- unique(unlist(top_200))
cat(paste0(length(union_top_200), "\n"))
mat <- data.frame(matrix(0, nrow = length(union_top_200), ncol = 48))
rownames(mat) <- union_top_200
colnames(mat) <- 1:48
for (i in 1:48) mat[top_200[[i]], i] <- 1
return(apply(mat, 1, sum))
}
agreement_wo_tree_filtering <- calculate_overlapping_candidates(data, validation_wo_tree_filtering)
agreement_w_tree_filtering <- calculate_overlapping_candidates(data, validation_w_tree_filtering)
y1 <- sapply(1:48, function(x) sum(agreement_wo_tree_filtering >= x))
y2 <- sapply(1:48, function(x) sum(agreement_w_tree_filtering >= x))
data_ <- data.frame(
x = rep(1:48, 2), y = c(y1, y2),
group = factor(c(rep(0, length(y1)), rep(1, length(y2))))
)
p4 <- ggplot(data_, aes(x = x, y = y, color = group)) +
geom_line(alpha = 0.7) +
scale_color_manual(values = c("#fc9d03", "red"), labels = c("Without tree filtering", "With tree filtering")) +
scale_x_reverse(breaks = c(48, 40, 30, 20, 10, 1)) +
scale_y_continuous(breaks = c(114, 150, 200, 250)) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(10, 5, 5, 5, "pt"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = c(0.72, 0.15),
legend.background = element_blank(),
legend.key = element_blank()
) +
xlab("Number of models") +
ylab("Number of candidate antigens")Final plot
p_combined <- plot_grid(p1, p2, p3, p4, nrow = 1, labels = c("a", "b", "c", "d"))
p_combined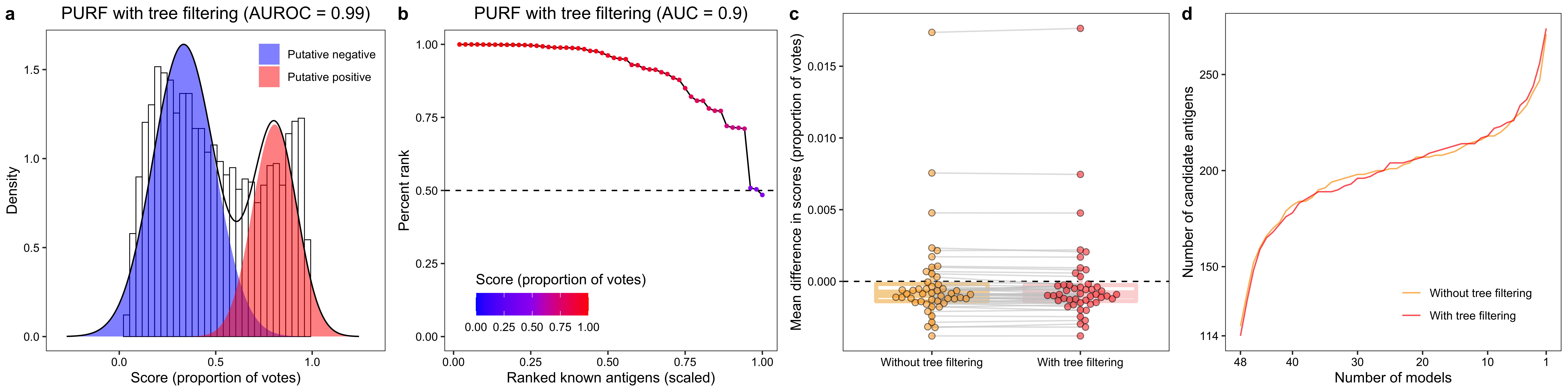
3.2 Model interpretation
PURF model interpretation with permutation-based variable importance and Wilcox test.
3.2.1 Analysis
3.2.1.1 Variable importance
Permutation-based variable importance and group variable importance.
In Python:
import pickle
import pandas as pd
import numpy as np
import pickle
from sklearn.impute import SimpleImputer
from sklearn.preprocessing import MinMaxScaler
from scipy.spatial import distance
import multiprocessing
from joblib import Parallel, delayed
num_cores = multiprocessing.cpu_count()
from sklearn.ensemble._forest import _generate_unsampled_indices
import session_infodata = pd.read_csv('./other_data/pf_ml_input_processed_weighted.csv', index_col=0)
pf3d7_features_processed = data.iloc[:,1:]
pf3d7_outcome = np.array(data.antigen_label)
purf_model = pickle.load(open('./pickle_data/0.5_purf_tree_filtering.pkl', 'rb'))
purf = purf_model['model']
weights = purf_model['weights']
metadata = pd.read_csv('./data/supplementary_data_2_pf_protein_variable_metadata.csv')
groups = metadata.loc[np.isin(metadata['column name'], pf3d7_features_processed.columns), 'category'].arraydef calculate_raw_var_imp_(idx, tree, X, y, weight, groups=None):
rng = np.random.RandomState(idx)
oob_indices = _generate_unsampled_indices(tree.random_state, y.shape[0], y.shape[0])
oob_pos = np.intersect1d(oob_indices, np.where(y == 1)[0])
noutall = len(oob_pos)
pred = tree.predict_proba(X.iloc[oob_pos,:])[:, 1]
nrightall = sum(pred == y[oob_pos])
imprt, impsd = [], []
if groups is None:
for var in range(X.shape[1]):
X_temp = X.copy()
X_temp.iloc[:, var] = rng.permutation(X_temp.iloc[:, var])
pred = tree.predict_proba(X_temp.iloc[oob_pos,:])[:, 1]
nrightimpall = sum(pred == y[oob_pos])
delta = (nrightall - nrightimpall) / noutall * weight
imprt.append(delta)
impsd.append(delta * delta)
else:
for grp in np.unique(groups):
X_temp = X.copy()
X_temp.iloc[:, groups == grp] = rng.permutation(X_temp.iloc[:, groups == grp])
pred = tree.predict_proba(X_temp.iloc[oob_pos,:])[:, 1]
nrightimpall = sum(pred == y[oob_pos])
delta = (nrightall - nrightimpall) / noutall * weight
imprt.append(delta)
impsd.append(delta * delta)
return (imprt, impsd)
def calculate_var_imp(model, features, outcome, num_cores, weights=None, groups=None):
trees = model.estimators_
idx_list = [i for i in range(len(trees))]
if weights is None:
weights = np.ones(len(trees))
res = Parallel(n_jobs=num_cores)(
delayed(calculate_raw_var_imp_)(idx, trees[idx], features, outcome, weights[idx], groups) for idx in idx_list)
imprt, impsd = [], []
for i in range(len(idx_list)):
imprt.append(res[i][0])
impsd.append(res[i][1])
imprt = np.array(imprt).sum(axis=0)
impsd = np.array(impsd).sum(axis=0)
imprt /= sum(weights)
impsd = np.sqrt(((impsd / sum(weights)) - imprt * imprt) / sum(weights))
mda = []
for i in range(len(imprt)):
if impsd[i] != 0:
mda.append(imprt[i] / impsd[i])
else:
mda.append(imprt[i])
if groups is None:
var_imp = pd.DataFrame({'variable': features.columns, 'meanDecreaseAccuracy': mda})
else:
var_imp = pd.DataFrame({'variable': np.unique(groups), 'meanDecreaseAccuracy': mda})
return var_impvar_imp = calculate_var_imp(purf, pf3d7_features_processed, pf3d7_outcome, num_cores, weights)
grp_var_imp = calculate_var_imp(purf, pf3d7_features_processed, pf3d7_outcome, num_cores, weights, groups)
var_imp.to_csv('./other_data/known_antigen_variable_importance.csv', index=False)
grp_var_imp.to_csv('./other_data/known_antigen_group_variable_importance.csv', index=False)3.2.1.2 Wilcoxon test
Variable value comparison between known antigens and other 52 random proteins predicted as negative.
In R:
library(stringr)
library(ggplot2)prediction <- read.csv("./data/supplementary_data_4_purf_oob_predictions.csv")
known_antigens <- prediction[prediction$antigen_label == 1, ]$protein_id
other_proteins <- prediction[prediction$antigen_label == 0 & prediction$oob_score_with_tree_filtering < 0.5, ]$protein_id
set.seed(22)
random_proteins <- sample(other_proteins, size = 52, replace = FALSE)
# Load imputed data
data <- read.csv("./other_data/pf_ml_input_processed_weighted.csv")
data <- data[!duplicated(data), ]
compared_group <- sapply(data$X, function(x) if (x %in% known_antigens) 1 else if (x %in% random_proteins) 0 else -1)
data <- data[, 3:ncol(data)]
# Min-max normalization
min_max <- function(x) {
(x - min(x)) / (max(x) - min(x))
}
data <- data.frame(lapply(data, min_max))
save(compared_group, data, file = "./rdata/known_antigen_wilcox_data.RData")pval <- c()
for (i in 1:ncol(data)) {
pval <- c(pval, wilcox.test(data[compared_group == 1, i], data[compared_group == 0, i])$p.value)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
# adj_pval = -log10(adj_pval)
wilcox_res <- data.frame(variable = colnames(data), adj_pval = adj_pval)
write.csv(wilcox_res, "./other_data/known_antigen_wilcox_res.csv", row.names = FALSE)3.2.2 Plotting
In R:
library(ggplot2)
library(reshape2)
library(cowplot)
library(stringr)
colorset <- c("genomic" = "#0C1C63", "immunological" = "#408002", "proteomic" = "#0F80FF", "structural" = "#FEAE34")3.2.2.1 Variable importance
var_imp <- read.csv("./other_data/known_antigen_variable_importance.csv")
var_imp <- var_imp[order(-var_imp$meanDecreaseAccuracy), ]
var_imp <- var_imp[1:10, ]
metadata <- read.csv("./data/supplementary_data_2_pf_protein_variable_metadata.csv", check.names = FALSE)
metadata <- metadata[c("category", "column name")]
metadata <- metadata[metadata$`column name` %in% var_imp$variable, ]
var_imp <- merge(x = var_imp, y = metadata, by.x = "variable", by.y = "column name")
var_imp$category <- factor(var_imp$category, levels = names(colorset))
var_imp$color <- sapply(var_imp$category, function(x) colorset[x])
var_imp_ <- var_imp
firstup <- function(x) {
substr(x, 1, 1) <- toupper(substr(x, 1, 1))
x
}
var_imp_$variable <- sapply(var_imp_$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
p1 <- ggplot(var_imp_, aes(x = reorder(variable, meanDecreaseAccuracy), y = meanDecreaseAccuracy, fill = category)) +
geom_point(size = 3, pch = 21, color = "black", alpha = 0.8) +
scale_fill_manual(values = colorset, labels = c("Genomic", "Immunological", "Proteomic", "Structural")) +
coord_flip() +
ylim(min(var_imp$meanDecreaseAccuracy), max(var_imp$meanDecreaseAccuracy) + 1) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.grid.major.y = element_line(color = "grey80", size = 0.3, linetype = "dotted"),
strip.background = element_blank(),
panel.border = element_rect(color = "black"),
legend.text = element_text(color = "black"),
plot.title = element_text(hjust = 0.5, size = 20),
plot.margin = ggplot2::margin(10, 10, 10, 10, "pt"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.title = element_blank(),
legend.position = c(0.7, 0.2),
legend.background = element_rect(colour = "black", size = 0.2)
) +
xlab("") +
ylab("Mean decrease in accuracy")3.2.2.2 Group variable importance
grp_var_imp <- read.csv("./other_data/known_antigen_group_variable_importance.csv")
grp_var_imp_ <- grp_var_imp
grp_var_imp_$variable <- sapply(grp_var_imp_$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
grp_var_imp_$category <- factor(tolower(grp_var_imp_$variable))
p2 <- ggplot(grp_var_imp_, aes(x = reorder(variable, meanDecreaseAccuracy), y = meanDecreaseAccuracy, fill = category)) +
geom_point(size = 3, pch = 21, color = "black", alpha = 0.8) +
scale_fill_manual(values = colorset, labels = c("Genomic", "Immunological", "Proteomic", "Structural")) +
coord_flip() +
ylim(min(grp_var_imp$meanDecreaseAccuracy), max(grp_var_imp$meanDecreaseAccuracy) + 5) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.grid.major.y = element_line(color = "grey80", size = 0.3, linetype = "dotted"),
strip.background = element_blank(),
panel.border = element_rect(color = "black"),
legend.text = element_text(color = "black", size = 10),
plot.title = element_text(hjust = 0.5, size = 20),
plot.margin = ggplot2::margin(10, 10, 10, 57, "pt"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.position = "none"
) +
xlab("") +
ylab("Mean decrease in accuracy")3.2.2.3 Wilcoxon test
load(file = "./rdata/known_antigen_wilcox_data.RData")
wilcox_res <- read.csv("./other_data/known_antigen_wilcox_res.csv")
wilcox_data <- data
wilcox_data$compared_group <- compared_group
wilcox_data <- wilcox_data[wilcox_data$compared_group != -1, ]
wilcox_data <- melt(wilcox_data, id = c("compared_group"))
wilcox_data <- merge(x = wilcox_data, y = merge(x = var_imp, y = wilcox_res), by = "variable", all.y = TRUE)
wilcox_data$tile_pos <- rep(0, nrow(wilcox_data))
wilcox_data$compared_group <- factor(wilcox_data$compared_group)
wilcox_data$variable <- sapply(wilcox_data$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
adj_pval_tmp <- c()
for (i in 1:nrow(wilcox_data)) {
x <- wilcox_data$adj_pval[i]
if (x >= 1e-3) {
res <- paste0("italic(p) == ", round(x, 3))
} else {
a <- strsplit(format(x, scientific = TRUE, digits = 3), "e")[[1]]
res <- paste0("italic(p) == ", as.numeric(a[1]), " %*% 10^", as.integer(a[2]))
}
adj_pval_tmp <- c(adj_pval_tmp, res)
}
wilcox_data$adj_pval <- adj_pval_tmp# p3_1 = ggplot(wilcox_data, aes(x=reorder(variable, meanDecreaseAccuracy), y=tile_pos)) +
# geom_text(aes(label=adj_pval), size=3, fontface='plain', family='sans', hjust=0, parse=TRUE) +
# scale_fill_gradient(low='blue', high='red') +
# coord_flip() +
# theme_bw() +
# xlab('') +
# ylab('') +
# ylim(0, 0.5)
#
# legend_1 = get_legend(p3_1 +
# theme(legend.title=element_text(vjust=0.7, color='black'),
# legend.background=element_blank()) +
# guides(fill=guide_colorbar(title=expression("-Log"[10]*"FDR"),
# direction = "horizontal")))
p3 <- ggplot(wilcox_data, aes(x = reorder(variable, meanDecreaseAccuracy), y = value, fill = compared_group)) +
geom_boxplot(outlier.color = NA, alpha = 0.3, lwd = 0.3) +
geom_point(
color = "black", shape = 21, stroke = 0.3, alpha = 0.5, size = 0.5,
position = position_jitterdodge()
) +
geom_text(aes(label = adj_pval), y = 1.1, size = 3, fontface = "plain", family = "sans", hjust = 0, parse = TRUE) +
geom_vline(xintercept = 1:9 + 0.5, color = "grey80", linetype = "solid", size = 0.1) +
coord_flip(ylim = c(0, 1), clip = "off") +
scale_fill_manual(
breaks = c("1", "0"), values = c("red", "blue"),
labels = c("Known antigens", "Random predicted non-antigens")
) +
theme_bw() +
xlab("") +
ylab("Normalized variable value")
legend_2 <- get_legend(p3 +
theme(
legend.title = element_blank(),
legend.background = element_blank(),
legend.key = element_blank(),
legend.direction = "horizontal",
legend.position = c(0.35, 0.9)
))
p3 <- p3 + theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
strip.background = element_blank(),
panel.border = element_rect(size = 0.2, colour = "black"),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(10, 90, 10, 0, "pt"),
legend.text = element_text(colour = "black"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.position = "none"
)Final plot
p_combined <- plot_grid(
plot_grid(p1, p3,
labels = c("a", "b"),
rel_widths = c(0.47, 0.53)
),
plot_grid(p2, NULL, legend_2,
labels = c("c", "", "", ""), nrow = 1,
rel_widths = c(0.47, 0.13, 0.4)
),
ncol = 1, rel_heights = c(0.65, 0.35)
)
p_combined
sessionInfo()## R version 4.2.3 (2023-03-15)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] stringr_1.4.1 ggpubr_0.4.0 ggbeeswarm_0.6.0 cowplot_1.1.1
## [5] ggplot2_3.4.2 rlist_0.4.6.2 pracma_2.3.8 mixR_0.2.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.9 lattice_0.20-45 tidyr_1.2.0 png_0.1-7
## [5] assertthat_0.2.1 digest_0.6.29 utf8_1.2.2 R6_2.5.1
## [9] backports_1.4.1 evaluate_0.16 highr_0.9 pillar_1.8.1
## [13] rlang_1.1.0 rstudioapi_0.14 data.table_1.14.2 car_3.1-0
## [17] jquerylib_0.1.4 R.utils_2.12.0 R.oo_1.25.0 Matrix_1.5-3
## [21] reticulate_1.25 rmarkdown_2.16 styler_1.8.0 munsell_0.5.0
## [25] broom_1.0.0 compiler_4.2.3 vipor_0.4.5 xfun_0.32
## [29] pkgconfig_2.0.3 htmltools_0.5.3 tidyselect_1.1.2 tibble_3.1.8
## [33] bookdown_0.28 codetools_0.2-19 fansi_1.0.3 dplyr_1.0.9
## [37] withr_2.5.0 R.methodsS3_1.8.2 jsonlite_1.8.0 gtable_0.3.0
## [41] lifecycle_1.0.3 DBI_1.1.3 magrittr_2.0.3 scales_1.2.1
## [45] carData_3.0-5 cli_3.6.1 stringi_1.7.8 cachem_1.0.6
## [49] ggsignif_0.6.3 bslib_0.4.0 generics_0.1.3 vctrs_0.6.2
## [53] tools_4.2.3 R.cache_0.16.0 glue_1.6.2 beeswarm_0.4.0
## [57] purrr_0.3.4 abind_1.4-5 fastmap_1.1.0 yaml_2.3.5
## [61] colorspace_2.0-3 rstatix_0.7.0 knitr_1.40 sass_0.4.2