Section 4 Candidate antigen clustering
4.1 Clustering analysis of the proximity matrix from the tree-filtered PURF model
4.1.1 Analysis
In Python:
import pandas as pd
import numpy as np
import pickle
from purf.pu_ensemble import PURandomForestClassifier
from sklearn.ensemble._forest import _generate_unsampled_indices
import multiprocessing
from joblib import Parallel, delayed
num_cores = multiprocessing.cpu_count()
import session_info# input set (5393, 1 + 272)
data = pd.read_csv('./data/supplementary_data_3_pf_ml_input.csv', index_col=0)
features = data.iloc[:, 1:]
outcome = np.array(data.antigen_label)
# Imputation
imputer = SimpleImputer(strategy='median')
X = imputer.fit_transform(features)
# Load model
model_tree_filtered = pickle.load(open('./pickle_data/0.5_purf_tree_filtering.pkl', 'rb'))
model = model_tree_filtered['model']
weights = model_tree_filtered['weights']
def calculate_proximity_(trees, y, leaf_indices):
same_leaf_mat = np.zeros([y.shape[0], y.shape[0]])
same_oob_mat = np.zeros([y.shape[0], y.shape[0]])
for idx, tree in enumerate(trees):
oob_indices = _generate_unsampled_indices(tree.random_state, y.shape[0], y.shape[0])
for i in oob_indices:
for j in oob_indices:
same_oob_mat[i, j] += 1
if leaf_indices[i, idx] == leaf_indices[j, idx]:
same_leaf_mat[i, j] += 1
return (same_leaf_mat, same_oob_mat)
# ------------------------------------------
# Calculate proximity without tree filtering
# ------------------------------------------
trees = model.estimators_
leaf_indices = model.apply(X)
idx_list = [i for i in range(len(trees))]
idx_list = np.array_split(idx_list, 1000)
res_1 = Parallel(n_jobs=num_cores)(
delayed(calculate_proximity_)(list(trees[i] for i in idx), outcome, leaf_indices[:, idx]) for idx in idx_list[0:round(len(idx_list) / 2)])
print('Done first part!')
res_2 = Parallel(n_jobs=num_cores)(
delayed(calculate_proximity_)(list(trees[i] for i in idx), outcome, leaf_indices[:, idx]) for idx in idx_list[round(len(idx_list) / 2):])
print('Done second part!')
same_leaf_mat = sum([r[0] for r in res_1 + res_2])
same_oob_mat = sum([r[1] for r in res_1 + res_2])
prox_mat = pd.DataFrame(np.divide(same_leaf_mat, same_oob_mat, out=np.zeros_like(same_leaf_mat),
where=same_oob_mat!=0))
prox_mat.index = features.index
prox_mat.columns = features.index
prox_mat.to_csv('./other_data/proximity_values.csv', index=False)
# ------------------------------------------
# Calculate proximity with tree filtering
# ------------------------------------------
trees = model.estimators_
leaf_indices = model.apply(X)
idx_list = [i for i in range(len(trees)) if weights[i] == 1]
idx_list = np.array_split(idx_list, 1000)
res = Parallel(n_jobs=num_cores)(
delayed(calculate_proximity_)(list(trees[i] for i in idx), outcome, leaf_indices[:, idx]) for idx in idx_list)
same_leaf_mat = sum([r[0] for r in res])
same_oob_mat = sum([r[1] for r in res])
prox_mat = pd.DataFrame(np.divide(same_leaf_mat, same_oob_mat, out=np.zeros_like(same_leaf_mat),
where=same_oob_mat!=0))
prox_mat.index = features.index
prox_mat.columns = features.index
prox_mat.to_csv('./other_data/proximity_values_tree_filtering.csv', index=False)4.1.1.1 Multi-dimensional scaling
In R:
# Multidimensional scaling
prox_mat <- read.csv("./other_data/proximity_values_tree_filtering.csv", check.names = FALSE)
mds <- cmdscale(as.dist(1 - prox_mat), k = ncol(prox_mat) - 1, eig = TRUE)
var_explained <- round(mds$eig * 100 / sum(mds$eig), 2)
print(paste0("Dimension 1 (", var_explained[1], "%)"))
print(paste0("Dimension 2 (", var_explained[2], "%)"))
print(paste0("Dimension 3 (", var_explained[3], "%)"))
mds <- as.data.frame(mds$points)
write.csv(mds, "./other_data/mds_proximity_values_tree_filtering.csv")4.1.2 Plotting
In R:
library(factoextra)
library(NbClust)
library(ggplot2)
library(stringr)
library(ggrepel)
library(cowplot)
library(reshape2)
library(rlist)
library(ggpubr)
library(ggbeeswarm)
library(ggforce)
library(cluster)
library(umap)prediction <- read.csv("./data/supplementary_data_4_purf_oob_predictions.csv")
prediction <- prediction[order(-prediction$oob_score_with_tree_filtering), ]
top_200 <- prediction[prediction$antigen_label == 0, ]$protein_id[1:200]
mds <- read.csv("./other_data/mds_proximity_values_tree_filtering.csv", row.names = 1, check.names = FALSE)
mds_ <- mds[rownames(mds) %in% top_200, ]4.1.2.1 Clustering on candidate antigens
# Gap statistic
set.seed(123)
gap_stat <- clusGap(mds_, kmeans, nstart = 100, K.max = 10, B = 100, iter.max = 50)
p0_1 <- fviz_gap_stat(gap_stat, maxSE = list(method = "Tibs2001SEmax", SE.factor = 2)) +
labs(title = "Gap statistic method")
# Silhouette method
set.seed(123)
p0_2 <- fviz_nbclust(mds_, kmeans, nstart = 100, method = "silhouette") +
labs(title = "Silhouette method")
# Elbow method
set.seed(123)
p0_3 <- fviz_nbclust(mds_, kmeans, nstart = 100, method = "wss") +
geom_vline(xintercept = 3, linetype = 2, color = "steelblue") +
labs(title = "Elbow method")label <- read.csv("./other_data/pf_antigen_labels.csv", row.names = 1, check.names = FALSE)
label$antigen_label[rownames(label) %in% c("PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1", "PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1")] <- 2
# Clustering
set.seed(123)
clusters <- kmeans(mds_, 3, nstart = 100)$cluster
label$antigen_label[rownames(label) %in% names(clusters)[clusters == 1]] <- -1
label$antigen_label[rownames(label) %in% names(clusters)[clusters == 2]] <- -2
label$antigen_label[rownames(label) %in% names(clusters)[clusters == 3]] <- -3
protein_id_1 <- rownames(label)[label$antigen_label == -1]
protein_id_2 <- rownames(label)[label$antigen_label == -2]
protein_id_3 <- rownames(label)[label$antigen_label == -3]
save(protein_id_1, protein_id_2, protein_id_3, file = "./rdata/clustering_groups.RData")# Save candidate gene accessions for GO enrichment analysis
gene_accession_1 <- str_replace_all(protein_id_1, "\\.[1-9]-p1", "")
gene_accession_2 <- str_replace_all(protein_id_2, "\\.[1-9]-p1", "")
gene_accession_3 <- str_replace_all(protein_id_3, "\\.[1-9]-p1", "")
write.table(data.frame("acc" = gene_accession_1),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_200_candidates_gene_accession_1.csv"
)
write.table(data.frame("acc" = gene_accession_2),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_200_candidates_gene_accession_2.csv"
)
write.table(data.frame("acc" = gene_accession_3),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_200_candidates_gene_accession_3.csv"
)set.seed(22)
umap_res <- umap(mds)
umap_df <- as.data.frame(umap_res$layout)
rownames(umap_df) <- rownames(mds)
umap_df$label <- label$antigen_label
umap_df_subset <- umap_df[umap_df$label %in% c(1, 2, -1, -2, -3), ]
umap_df_subset$label <- factor(umap_df_subset$label)
umap_df_subset <- umap_df_subset[order(umap_df_subset$label), ]
umap_df_subset$label_text <- ""
umap_df_subset$label_text[rownames(umap_df_subset) == "PF3D7_0304600.1-p1"] <- "CSP"
umap_df_subset$label_text[rownames(umap_df_subset) == "PF3D7_0424100.1-p1"] <- "RH5"
umap_df_subset$label_text[rownames(umap_df_subset) == "PF3D7_0206900.1-p1"] <- "MSP5"
umap_df_subset$label_text[rownames(umap_df_subset) == "PF3D7_0209000.1-p1"] <- "P230"p1_1 <- ggplot(data = umap_df_subset, aes(x = `V1`, y = `V2`)) +
geom_point(aes(fill = label), shape = 21, alpha = 0.7, color = "grey30") +
# geom_mark_ellipse(aes(fill=label, filter=!label %in% c(1,2)), size=0, alpha=0.2, expand=unit(1, "mm"), color='grey90') +
scale_fill_manual(breaks = c(-1, -2, -3, 1, 2), values = c("#03a1fc", "#984EA3", "#FF7F00", "#b8ffe0", "#ffee00"), labels = c("Candidate antigen (Group 1)", "Candidate antigen (Group 2)", "Candidate antigen (Group 3)", "Known antigen", "Reference antigen")) +
geom_text_repel(aes(label = label_text), size = 3.5, nudge_x = -0.5, nudge_y = 1, point.padding = 0.1) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 0, 0, 5, "pt"),
axis.title = element_text(colour = "black"),
axis.text.x = element_text(colour = "black"),
axis.text.y = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_blank(),
legend.background = element_blank(),
legend.key = element_blank()
) +
xlim(min(umap_df_subset$`V1`), max(umap_df_subset$`V1`)) +
ylim(min(umap_df_subset$`V2`), max(umap_df_subset$`V2`)) +
xlab("Dimension 1") +
ylab("Dimension 2")4.1.2.2 Difference in scores
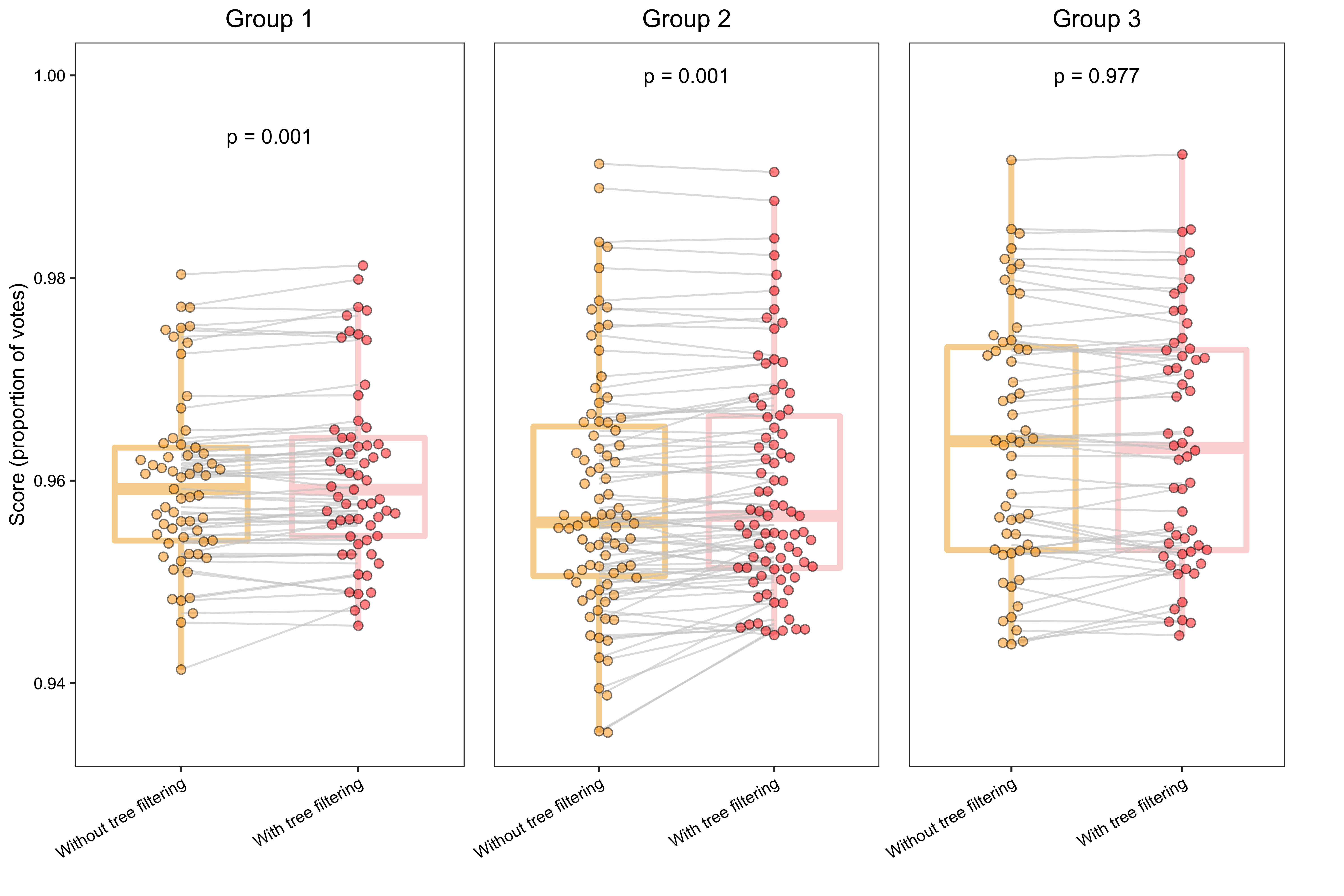
Prediction scores of clustering groups before and after tree filtering.
prediction <- read.csv("./data/supplementary_data_4_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)[, 2:3]
load(file = "./rdata/clustering_groups.RData")
data_subset <- prediction[rownames(prediction) %in% c(protein_id_1, protein_id_2, protein_id_3), ]
data_subset$label <- 0
data_subset$label[rownames(data_subset) %in% protein_id_1] <- "Group 1"
data_subset$label[rownames(data_subset) %in% protein_id_2] <- "Group 2"
data_subset$label[rownames(data_subset) %in% protein_id_3] <- "Group 3"
data_subset$label <- factor(data_subset$label)pval <- c()
ds <- list()
for (i in 1:3) {
ds_ <- melt(data_subset[data_subset$label == paste0("Group ", i), ])
ds_$paired <- rep(1:(nrow(ds_) / 2), 2)
pval <- c(pval, compare_means(value ~ variable, data = ds_, method = "wilcox.test", paired = TRUE)$p)
ds <- list.append(ds, ds_)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
p2_1 <- ggplot(data = ds[[1]], aes(x = variable, y = value)) +
geom_boxplot(aes(color = variable), outlier.color = NA, lwd = 1.5, show.legend = TRUE) +
geom_line(aes(group = paired), alpha = 0.6, color = "grey80") +
geom_beeswarm(aes(fill = variable),
color = "black", alpha = 0.5, size = 2, cex = 2.5, priority = "random",
shape = 21
) +
# annotate("segment", x=1, xend=2, y=0.991, yend=0.991, colour="black") +
# annotate("segment", x=1, xend=1, y=0.991, yend=0.989, colour="black") +
# annotate("segment", x=2, xend=2, y=0.991, yend=0.989, colour="black") +
annotate("text", x = 1.5, y = 0.994, label = paste0("p = ", round(adj_pval[1], 3))) +
scale_color_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#f7d59e", "#fcd7d7"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_fill_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#fc9d03", "red"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_x_discrete(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
labels = c("Without tree filtering", "With tree filtering")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 0, 5, 5, "pt"),
axis.title = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
axis.text.x = element_text(angle = 30, colour = "black", hjust = 1, vjust = 1),
axis.text.y = element_text(colour = "black"),
legend.position = "none"
) +
xlab("") +
ylab("Score (proportion of votes)") +
ylim(0.935, 1) +
ggtitle("Group 1")
p2_2 <- ggplot(data = ds[[2]], aes(x = variable, y = value)) +
geom_boxplot(aes(color = variable), outlier.color = NA, lwd = 1.5, show.legend = TRUE) +
geom_line(aes(group = paired), alpha = 0.6, color = "grey80") +
geom_beeswarm(aes(fill = variable),
color = "black", alpha = 0.5, size = 2, cex = 2.5, priority = "random",
shape = 21
) +
# annotate("segment", x=1, xend=2, y=0.997, yend=0.997, colour="black") +
# annotate("segment", x=1, xend=1, y=0.997, yend=0.995, colour="black") +
# annotate("segment", x=2, xend=2, y=0.997, yend=0.995, colour="black") +
annotate("text", x = 1.5, y = 1, label = paste0("p = ", round(adj_pval[2], 3))) +
scale_color_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#f7d59e", "#fcd7d7"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_fill_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#fc9d03", "red"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_x_discrete(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
labels = c("Without tree filtering", "With tree filtering")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 0, 5, 0, "pt"),
axis.title = element_text(colour = "black"),
axis.text.x = element_text(angle = 30, colour = "black", hjust = 1, vjust = 1),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
) +
xlab("") +
ylab("") +
ylim(0.935, 1) +
ggtitle("Group 2")
p2_3 <- ggplot(data = ds[[3]], aes(x = variable, y = value)) +
geom_boxplot(aes(color = variable), outlier.color = NA, lwd = 1.5, show.legend = TRUE) +
geom_line(aes(group = paired), alpha = 0.6, color = "grey80") +
geom_beeswarm(aes(fill = variable),
color = "black", alpha = 0.5, size = 2, cex = 2.5, priority = "random",
shape = 21
) +
# annotate("segment", x=1, xend=2, y=0.997, yend=0.997, colour="black") +
# annotate("segment", x=1, xend=1, y=0.997, yend=0.995, colour="black") +
# annotate("segment", x=2, xend=2, y=0.997, yend=0.995, colour="black") +
annotate("text", x = 1.5, y = 1, label = paste0("p = ", round(adj_pval[3], 3))) +
scale_color_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#f7d59e", "#fcd7d7"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_fill_manual(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
values = c("#fc9d03", "red"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_x_discrete(
breaks = c(
"oob_score_without_tree_filtering",
"oob_score_with_tree_filtering"
),
labels = c("Without tree filtering", "With tree filtering")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 5, 5, 0, "pt"),
axis.title = element_text(colour = "black"),
axis.text.x = element_text(angle = 30, colour = "black", hjust = 1, vjust = 1),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
) +
xlab("") +
ylab("") +
ylim(0.935, 1) +
ggtitle("Group 3")Final plot
# p_1 = plot_grid(p1_1, legend_1, p1_2, p1_3, nrow=2, rel_widths=c(0.5, 0.5))
p_2 <- plot_grid(p2_1, p2_2, p2_3, NULL, nrow = 1, rel_widths = c(0.28, 0.25, 0.25, 0.03))png(file = "./figures/Fig 4.png", width = 3500, height = 2200, res = 600)
print(p1_1)
dev.off()
pdf(file = "../figures/Fig 4.pdf", width = 7, height = 4.4)
print(p1_1)
dev.off()
png(file = "./figures/Supplementary Fig 8.png", width = 6000, height = 4000, res = 600)
print(p_2)
dev.off()
pdf(file = "../supplementary_figures/Supplementary Fig 8.pdf", width = 12, height = 8)
print(p_2)
dev.off()
4.2 Comparison of the candidate clustering groups
Comparisons of clustering groups before and after tree filtering in terms of distances to reference antigens and predictions scores.
4.2.1 Plotting
In R:
library(ggplot2)
library(ggbeeswarm)
library(cowplot)
library(ggpubr)
library(rlist)prox_wo_tree_filtering <- read.csv("./other_data/proximity_values.csv", check.names = FALSE)
prox_w_tree_filtering <- read.csv("./other_data/proximity_values_tree_filtering.csv", check.names = FALSE)
rownames(prox_wo_tree_filtering) <- colnames(prox_wo_tree_filtering)
rownames(prox_w_tree_filtering) <- colnames(prox_w_tree_filtering)
dist_wo_tree_filtering <- 1 - prox_wo_tree_filtering
dist_w_tree_filtering <- 1 - prox_w_tree_filtering
diff_dist <- dist_w_tree_filtering - dist_wo_tree_filtering
load(file = "./rdata/clustering_groups.RData")# CSP, RH5, MSP5, P230
ref <- c("PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1", "PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1")
dist_wo_tree_filtering[ref, ref]
dist_w_tree_filtering[ref, ref]
diff_dist[ref, ref]plot_boxplots <- function(protein_id, protein_name, ylim1, ylim2) {
dist_0 <- dist_wo_tree_filtering[c(protein_id_1, protein_id_2, protein_id_3), protein_id]
dist_1 <- dist_w_tree_filtering[c(protein_id_1, protein_id_2, protein_id_3), protein_id]
dist_smry <- data.frame(
label = rep(c(
rep("Group 1", length(protein_id_1)),
rep("Group 2", length(protein_id_2)),
rep("Group 3", length(protein_id_3))
)),
value = c(dist_0, dist_1),
variable = factor(c(rep(0, length(dist_0)), rep(1, length(dist_1))))
)
pval <- c()
ds <- list()
for (i in 1:3) {
ds_ <- dist_smry[dist_smry$label == paste0("Group ", i), ]
ds_$paired <- rep(1:(nrow(ds_) / 2), 2)
pval <- c(pval, compare_means(value ~ variable,
data = ds_, method = "wilcox.test",
paired = TRUE
)$p)
ds <- list.append(ds, ds_)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
plots <- list()
for (i in 1:3) {
p <- ggplot(data = ds[[i]], aes(x = variable, y = value)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "grey60") +
geom_boxplot(aes(color = variable), outlier.color = NA, lwd = 1.5, show.legend = FALSE, fill = "transparent") +
geom_line(aes(group = paired), alpha = 0.6, color = "grey80") +
geom_beeswarm(aes(fill = variable),
color = "black", alpha = 0.5, size = 2, cex = 2,
priority = "random", shape = 21
) +
# annotate("segment", x=1, xend=2, y=max(ds[[i]]$value) + 0.04, yend=max(ds[[i]]$value) + 0.04,
# colour="black") +
# annotate("segment", x=1, xend=1, y=max(ds[[i]]$value) + 0.04, yend=max(ds[[i]]$value) + 0.03,
# colour="black") +
# annotate("segment", x=2, xend=2, y=max(ds[[i]]$value) + 0.04, yend=max(ds[[i]]$value) + 0.03,
# colour="black") +
{
if (adj_pval[i] >= 1e-3) {
annotate("text",
x = 1.5, y = max(ds[[i]]$value) + 0.06,
label = paste0("p = ", round(adj_pval[i], 3))
)
} else {
annotate("text",
x = 1.5, y = max(ds[[i]]$value) + 0.06,
label = paste0("p = ", formatC(adj_pval[i], format = "e", digits = 2))
)
}
} +
scale_color_manual(values = c("#f7d59e", "#fcd7d7")) +
scale_fill_manual(
values = c("#fc9d03", "red"),
labels = c("Without tree filtering", "With tree filtering")
) +
scale_x_discrete(labels = c("Without tree filtering", "With tree filtering")) +
theme_bw() +
{
if (i == 1) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 0, 5, 25, "pt"),
axis.title = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black", size = 8),
axis.text.x = element_text(colour = "black", angle = 45, vjust = 1, hjust = 1),
axis.text.y = element_text(colour = "black"),
rect = element_rect(fill = "transparent"),
legend.position = "none"
)
} else if (i == 2) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 0, 5, -5, "pt"),
axis.title = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black", size = 8),
axis.text.x = element_text(colour = "black", angle = 45, vjust = 1, hjust = 1),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
rect = element_rect(fill = "transparent"),
legend.position = "none"
)
} else {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 5, 5, -5, "pt"),
axis.title = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black", size = 8),
axis.text.x = element_text(colour = "black", angle = 45, vjust = 1, hjust = 1),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
rect = element_rect(fill = "transparent"),
legend.position = "none"
)
}
} +
xlab("") +
{
if (i == 1) {
ylab(paste0("Euclidean distance to ", protein_name))
} else {
ylab("")
}
} +
ylim(ylim1) +
ggtitle(paste0("Group ", i))
plots <- list.append(plots, p)
}
# Plot difference
diff_grps <- diff_dist[c(protein_id_1, protein_id_2, protein_id_3), protein_id]
ds <- data.frame(
value = c(diff_grps),
group = factor(c(
rep("Group 1", length(protein_id_1)),
rep("Group 2", length(protein_id_2)),
rep("Group 3", length(protein_id_3))
))
)
stats <- compare_means(value ~ group, data = ds, method = "wilcox.test", p.adjust.method = "BH")
p <- ggplot(data = ds, aes(x = group, y = value, fill = group)) +
geom_hline(yintercept = 0, linetype = "dashed", color = "grey80") +
geom_boxplot(outlier.colour = NA, alpha = 0.3, lwd = 0.3) +
geom_beeswarm(aes(fill = group),
color = "black", alpha = 0.5, size = 1.5, cex = 1.5,
priority = "random", shape = 21
) +
annotate("segment", x = 1, xend = 1.95, y = max(ds$value) + 0.004, yend = max(ds$value) + 0.004, colour = "black") +
annotate("segment", x = 1, xend = 1, y = max(ds$value) + 0.002, yend = max(ds$value) + 0.004, colour = "black") +
annotate("segment", x = 1.95, xend = 1.95, y = max(ds$value) + 0.002, yend = max(ds$value) + 0.004, colour = "black") +
annotate("text", x = 1.5, y = max(ds$value) + 0.007, label = paste0("p = ", stats$p.adj[1])) +
annotate("segment", x = 1, xend = 3, y = max(ds$value) + 0.01, yend = max(ds$value) + 0.01, colour = "black") +
annotate("segment", x = 1, xend = 1, y = max(ds$value) + 0.008, yend = max(ds$value) + 0.01, colour = "black") +
annotate("segment", x = 3, xend = 3, y = max(ds$value) + 0.008, yend = max(ds$value) + 0.01, colour = "black") +
annotate("text", x = 2, y = max(ds$value) + 0.013, label = paste0("p = ", stats$p.adj[2])) +
annotate("segment", x = 2.05, xend = 3, y = max(ds$value) + 0.004, yend = max(ds$value) + 0.004, colour = "black") +
annotate("segment", x = 2.05, xend = 2.05, y = max(ds$value) + 0.002, yend = max(ds$value) + 0.004, colour = "black") +
annotate("segment", x = 3, xend = 3, y = max(ds$value) + 0.002, yend = max(ds$value) + 0.004, colour = "black") +
annotate("text", x = 2.5, y = max(ds$value) + 0.007, label = paste0("p = ", stats$p.adj[3])) +
scale_fill_manual(
breaks = c("Group 1", "Group 2", "Group 3"),
values = c("#03a1fc", "#984EA3", "#FF7F00"),
labels = c(
"Candidate antigen (Group 1)",
"Candidate antigen (Group 2)",
"Candidate antigen (Group 3)"
)
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 5, 38, 15, "pt"),
axis.title = element_text(colour = "black"),
axis.text.x = element_text(colour = "black", angle = 45, vjust = 1, hjust = 1),
axis.text.y = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
rect = element_rect(fill = "transparent"),
legend.position = "none"
) +
xlab("") +
ylab(paste0("Difference in distances to ", protein_name)) +
ylim(ylim2) +
ggtitle("")
plots <- list.append(plots, p)
return(plots)
}p1 <- plot_grid(ggdraw() + draw_label("CSP (circumsporozoite protein); pre-erythrocytic stage"),
plot_grid(
plotlist = plot_boxplots(
protein_id = "PF3D7_0304600.1-p1", protein_name = "CSP",
ylim1 = c(0.15, 0.7), ylim2 = c(-0.03, 0.04)
),
nrow = 1, rel_widths = c(0.29, 0.22, 0.23, 0.48)
),
ncol = 1, rel_heights = c(0.1, 1), labels = c("a", "")
)
p2 <- plot_grid(ggdraw() + draw_label("RH5 (reticulocyte binding protein homologue 5); erythrocytic stage"),
plot_grid(
plotlist = plot_boxplots(
protein_id = "PF3D7_0424100.1-p1", protein_name = "RH5",
ylim1 = c(0.15, 0.7), ylim2 = c(-0.03, 0.04)
),
nrow = 1, rel_widths = c(0.29, 0.22, 0.23, 0.48)
),
ncol = 1, rel_heights = c(0.1, 1), labels = c("b", "")
)
p3 <- plot_grid(ggdraw() + draw_label("MSP5 (merozoite surface protein 5); erythrocytic stage"),
plot_grid(
plotlist = plot_boxplots(
protein_id = "PF3D7_0206900.1-p1", protein_name = "MSP5",
ylim1 = c(0.15, 0.7), ylim2 = c(-0.03, 0.04)
),
nrow = 1, rel_widths = c(0.29, 0.22, 0.23, 0.48)
),
ncol = 1, rel_heights = c(0.1, 1), labels = c("c", "")
)
p4 <- plot_grid(ggdraw() + draw_label("P230 (6-cysteine protein); gametocyte stage"),
plot_grid(
plotlist = plot_boxplots(
protein_id = "PF3D7_0209000.1-p1", protein_name = "P230",
ylim1 = c(0.15, 0.7), ylim2 = c(-0.03, 0.04)
),
nrow = 1, rel_widths = c(0.29, 0.22, 0.23, 0.48)
),
ncol = 1, rel_heights = c(0.1, 1), labels = c("d", "")
)Final plot
p_combined <- plot_grid(p1, p2, p3, p4, nrow = 2)
p_combined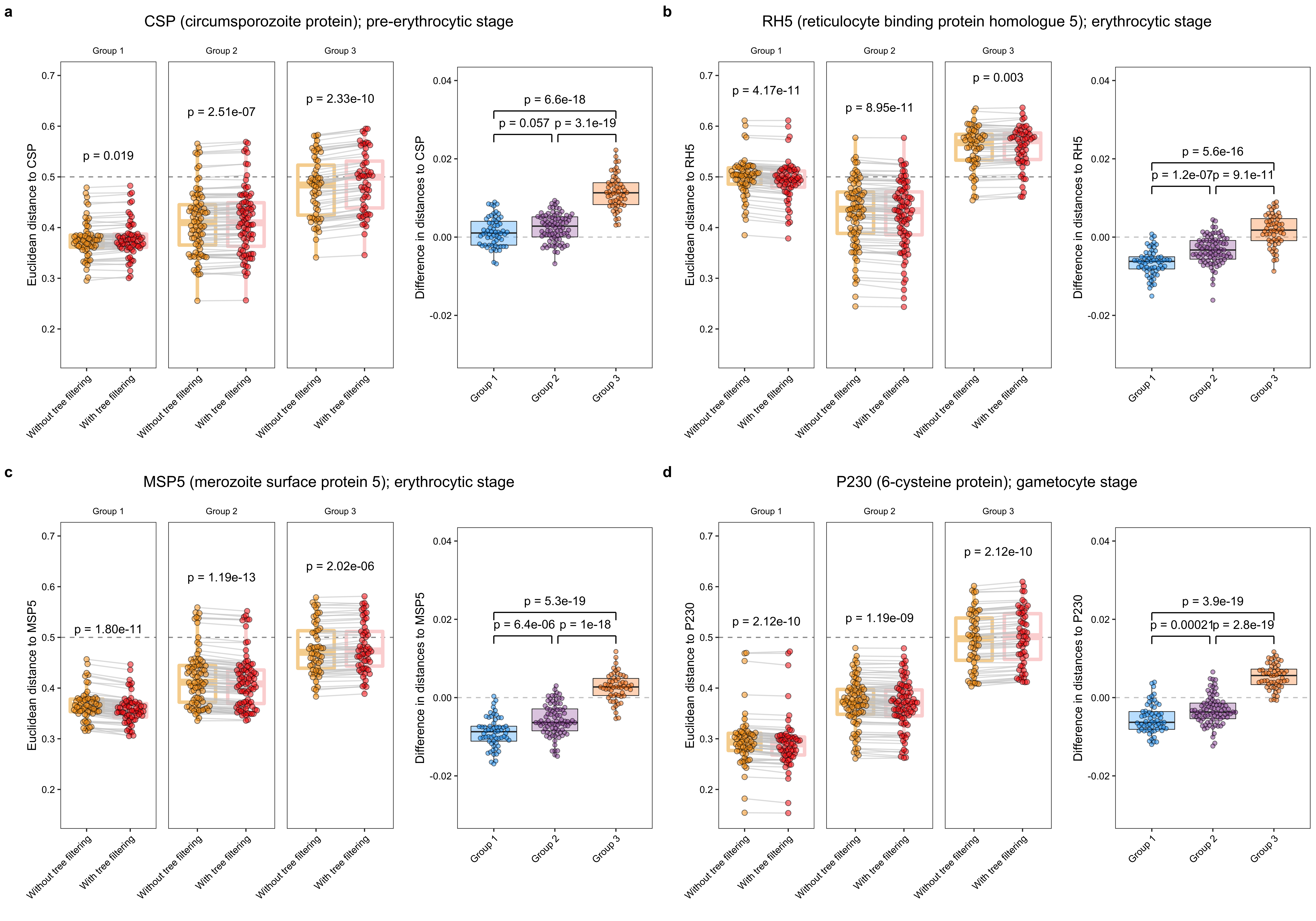
4.3 Important variables for candidate clustering groups
Wilcox test to compare variable values in candidate groups and randomly selected proteins.
4.3.1 Analysis
Permutation-based variable importance of candidate antigen groups.
4.3.1.1 Variable importance
In R:
data <- read.csv("./other_data/pf_ml_input_processed_weighted.csv")
load(file = "./rdata/clustering_groups.RData")
data_1 <- data
data_1$antigen_label <- 0
data_1$antigen_label[data_1$X %in% protein_id_1] <- 1
write.csv(data_1, "./other_data/pf_ml_input_processed_weighted_group_1.csv", row.names = FALSE)
data_2 <- data
data_2$antigen_label <- 0
data_2$antigen_label[data_2$X %in% protein_id_2] <- 1
write.csv(data_2, "./other_data/pf_ml_input_processed_weighted_group_2.csv", row.names = FALSE)
data_3 <- data
data_3$antigen_label <- 0
data_3$antigen_label[data_3$X %in% protein_id_3] <- 1
write.csv(data_3, "./other_data/pf_ml_input_processed_weighted_group_3.csv", row.names = FALSE)In Python:
import pickle
import pandas as pd
import numpy as np
import pickle
from sklearn.impute import SimpleImputer
from sklearn.preprocessing import MinMaxScaler
from scipy.spatial import distance
import multiprocessing
from joblib import Parallel, delayed
num_cores = multiprocessing.cpu_count()
from sklearn.ensemble._forest import _generate_unsampled_indices
import session_infodata_1 = pd.read_csv('./other_data/pf_ml_input_processed_weighted_group_1.csv', index_col=0)
pf3d7_features_processed_1 = data_1.iloc[:,1:].apply(pd.to_numeric, downcast='float')
pf3d7_outcome_1 = np.array(data_1.antigen_label)
data_2 = pd.read_csv('./other_data/pf_ml_input_processed_weighted_group_2.csv', index_col=0)
pf3d7_features_processed_2 = data_2.iloc[:,1:].apply(pd.to_numeric, downcast='float')
pf3d7_outcome_2 = np.array(data_2.antigen_label)
data_3 = pd.read_csv('./other_data/pf_ml_input_processed_weighted_group_3.csv', index_col=0)
pf3d7_features_processed_3 = data_3.iloc[:,1:].apply(pd.to_numeric, downcast='float')
pf3d7_outcome_3 = np.array(data_3.antigen_label)
purf_model = pickle.load(open('./pickle_data/pf_purf_tree_filtering.pkl', 'rb'))
purf = purf_model['model']
weights = purf_model['weights']
metadata = pd.read_csv('./data/supplementary_data_2_pf_protein_variable_metadata.csv')def calculate_raw_var_imp_(idx, tree, X, y, weight, groups=None):
rng = np.random.RandomState(idx)
oob_indices = _generate_unsampled_indices(tree.random_state, y.shape[0], y.shape[0])
oob_pos = np.intersect1d(oob_indices, np.where(y == 1)[0])
noutall = len(oob_pos)
pred = tree.predict_proba(X.iloc[oob_pos,:])[:, 1]
nrightall = sum(pred == y[oob_pos])
imprt, impsd = [], []
if groups is None:
for var in range(X.shape[1]):
X_temp = X.copy().iloc[oob_pos,:]
X_temp.iloc[:, var] = rng.permutation(X_temp.iloc[:, var])
pred = tree.predict_proba(X_temp)[:, 1]
nrightimpall = sum(pred == y[oob_pos])
delta = (nrightall - nrightimpall) / noutall * weight
imprt.append(delta)
impsd.append(delta * delta)
else:
for grp in np.unique(groups):
X_temp = X.copy().iloc[oob_pos,:]
X_temp.iloc[:, groups == grp] = rng.permutation(X_temp.iloc[:, groups == grp])
pred = tree.predict_proba(X_temp)[:, 1]
nrightimpall = sum(pred == y[oob_pos])
delta = (nrightall - nrightimpall) / noutall * weight
imprt.append(delta)
impsd.append(delta * delta)
return (imprt, impsd)
def calculate_var_imp(model, features, outcome, num_cores, weights=None, groups=None):
trees = model.estimators_
idx_list = [i for i in range(len(trees))]
if weights is None:
weights = np.ones(len(trees))
res = Parallel(n_jobs=num_cores)(
delayed(calculate_raw_var_imp_)(idx, trees[idx], features, outcome, weights[idx], groups) for idx in idx_list)
imprt, impsd = [], []
for i in range(len(idx_list)):
imprt.append(res[i][0])
impsd.append(res[i][1])
imprt = np.array(imprt).sum(axis=0)
impsd = np.array(impsd).sum(axis=0)
imprt /= len(idx_list)
impsd = np.sqrt(((impsd / len(idx_list)) - imprt * imprt) / len(idx_list))
mda = []
for i in range(len(imprt)):
if impsd[i] != 0:
mda.append(imprt[i] / impsd[i])
else:
mda.append(imprt[i])
if groups is None:
var_imp = pd.DataFrame({'variable': features.columns, 'meanDecreaseAccuracy': mda})
else:
var_imp = pd.DataFrame({'variable': np.unique(groups), 'meanDecreaseAccuracy': mda})
return var_impvar_imp_1 = calculate_var_imp(purf, pf3d7_features_processed_1, pf3d7_outcome_1, num_cores, weights)
var_imp_1.to_csv('./other_data/candidate_variable_importance_1.csv', index=False)
var_imp_2 = calculate_var_imp(purf, pf3d7_features_processed_2, pf3d7_outcome_2, num_cores, weights)
var_imp_2.to_csv('./other_data/candidate_variable_importance_2.csv', index=False)
var_imp_3 = calculate_var_imp(purf, pf3d7_features_processed_3, pf3d7_outcome_3, num_cores, weights)
var_imp_3.to_csv('./other_data/candidate_variable_importance_3.csv', index=False)4.3.1.2 Wilcoxon test
Variable value comparison between candidate antigens and other random proteins predicted as negative.
In R:
library(stringr)
library(ggplot2)prediction <- read.csv("./data/supplementary_data_4_purf_oob_predictions.csv")
load(file = "./rdata/clustering_groups.RData")
other_proteins <- prediction[prediction$antigen_label == 0 & prediction$oob_score_with_tree_filtering < 0.5, ]$protein_id
set.seed(22)
random_protein_1 <- sample(other_proteins, size = length(protein_id_1), replace = FALSE)
random_protein_2 <- sample(other_proteins, size = length(protein_id_2), replace = FALSE)
random_protein_3 <- sample(other_proteins, size = length(protein_id_3), replace = FALSE)
# Load imputed data
data <- read.csv("./other_data/pf_ml_input_processed_weighted.csv")
data <- data[!duplicated(data), ]
compared_group_1 <- sapply(data$X, function(x) if (x %in% protein_id_1) 1 else if (x %in% random_protein_1) 0 else -1)
compared_group_2 <- sapply(data$X, function(x) if (x %in% protein_id_2) 1 else if (x %in% random_protein_2) 0 else -1)
compared_group_3 <- sapply(data$X, function(x) if (x %in% protein_id_3) 1 else if (x %in% random_protein_3) 0 else -1)
data <- data[, 3:ncol(data)]
# Min-max normalization
min_max <- function(x) {
(x - min(x)) / (max(x) - min(x))
}
data <- data.frame(lapply(data, min_max))
save(compared_group_1, compared_group_2, compared_group_3, data, file = "./rdata/candidate_antigen_wilcox_data.RData")pval <- c()
for (i in 1:ncol(data)) {
pval <- c(pval, wilcox.test(data[compared_group_1 == 1, i], data[compared_group_1 == 0, i])$p.value)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
# adj_pval = -log10(adj_pval)
wilcox_res <- data.frame(variable = colnames(data), adj_pval = adj_pval)
write.csv(wilcox_res, "./other_data/candidate_antigen_wilcox_res_1.csv", row.names = FALSE)
pval <- c()
for (i in 1:ncol(data)) {
pval <- c(pval, wilcox.test(data[compared_group_2 == 1, i], data[compared_group_2 == 0, i])$p.value)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
# adj_pval = -log10(adj_pval)
wilcox_res <- data.frame(variable = colnames(data), adj_pval = adj_pval)
write.csv(wilcox_res, "./other_data/candidate_antigen_wilcox_res_2.csv", row.names = FALSE)
pval <- c()
for (i in 1:ncol(data)) {
pval <- c(pval, wilcox.test(data[compared_group_3 == 1, i], data[compared_group_3 == 0, i])$p.value)
}
adj_pval <- p.adjust(pval, method = "BH", n = length(pval))
# adj_pval = -log10(adj_pval)
wilcox_res <- data.frame(variable = colnames(data), adj_pval = adj_pval)
write.csv(wilcox_res, "./other_data/candidate_antigen_wilcox_res_3.csv", row.names = FALSE)4.3.2 Plotting
In R:
library(ggplot2)
library(reshape2)
library(cowplot)
library(sfsmisc)
library(stringr)firstup <- function(x) {
substr(x, 1, 1) <- toupper(substr(x, 1, 1))
x
}
load(file = "./rdata/candidate_antigen_wilcox_data.RData")wilcox_res <- read.csv("./other_data/candidate_antigen_wilcox_res_1.csv")
wilcox_data <- data
wilcox_data$compared_group <- compared_group_1
var_imp <- read.csv("./other_data/processed_candidate_variable_importance_1.csv", row.names = 1, check.names = FALSE)
wilcox_data <- wilcox_data[wilcox_data$compared_group != -1, ]
wilcox_data <- melt(wilcox_data, id = c("compared_group"))
wilcox_data <- merge(x = wilcox_data, y = merge(x = var_imp, y = wilcox_res, by.x = "row.names", by.y = "variable"), by.x = "variable", by.y = "Row.names", all.y = TRUE)
wilcox_data$tile_pos <- rep(0, nrow(wilcox_data))
wilcox_data$compared_group <- factor(wilcox_data$compared_group)
wilcox_data$variable <- sapply(wilcox_data$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
adj_pval_tmp <- c()
for (i in 1:nrow(wilcox_data)) {
x <- wilcox_data$adj_pval[i]
if (x >= 1e-3) {
res <- paste0("italic(p) == ", round(x, 3))
} else {
a <- strsplit(format(x, scientific = TRUE, digits = 2), "e")[[1]]
res <- paste0("italic(p) == ", as.numeric(a[1]), " %*% 10^", as.integer(a[2]))
}
adj_pval_tmp <- c(adj_pval_tmp, res)
}
wilcox_data$adj_pval <- adj_pval_tmp
p1 <- ggplot(wilcox_data, aes(x = reorder(variable, `Mean decrease accuracy`), y = value, fill = compared_group)) +
geom_boxplot(outlier.color = NA, alpha = 0.3, lwd = 0.3) +
geom_point(
color = "black", shape = 21, stroke = 0.3, alpha = 0.5, size = 0.5,
position = position_jitterdodge()
) +
geom_text(aes(label = adj_pval), y = 1.1, size = 3, fontface = "plain", family = "sans", hjust = 0, parse = TRUE) +
geom_vline(xintercept = 1:9 + 0.5, color = "grey80", linetype = "solid", size = 0.1) +
scale_x_discrete(
breaks = sapply(c(
"nonsynonymous_snps",
"max_karplus_schulz_flexibility",
"max_parker_hydrophilicity",
"PmN",
"mitochondrial",
"avg_parker_hydrophilicity",
"hydrophobicity_Group3",
"total_snps",
"min_score_ifn_epitopes",
"pi_value"
), function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
}),
labels = c(
"Number non-synonymous SNPs",
"Maximum score of Karplus and Schulz flexibility",
"Maximum score of Parker hydrophilicity",
expression("% positive charge a.a. - % negative charge a.a."),
"Mitochondrial",
"Average score of Parker hydrophilicity",
expression("% hydrophobicity amino acids"),
"Total number of SNPs",
"Minimum score of IFN-gamma inducing epitopes",
"Isoelectric point (PI) value"
)
) +
coord_flip(ylim = c(0, 1), clip = "off") +
scale_fill_manual(
breaks = c("1", "0"), values = c("red", "blue"),
labels = c("Candidate antigens", "Random predicted non-antigens")
) +
theme_bw() +
xlab("") +
ylab("Normalized variable value") +
ylim(0, 1) +
ggtitle("Group 1 (61 candidates)")
legend <- get_legend(p1 + theme(
legend.title = element_blank(),
legend.background = element_blank(),
legend.key = element_blank(),
legend.direction = "horizontal",
legend.position = c(0.35, 0.9)
))
p1 <- p1 + theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
strip.background = element_blank(),
panel.border = element_rect(colour = "black"),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(10, 70, 5, 30, "pt"),
legend.text = element_text(colour = "black"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.position = "none"
)wilcox_res <- read.csv("./other_data/candidate_antigen_wilcox_res_2.csv")
wilcox_data <- data
wilcox_data$compared_group <- compared_group_2
var_imp <- read.csv("./other_data/processed_candidate_variable_importance_2.csv", row.names = 1, check.names = FALSE)
wilcox_data <- wilcox_data[wilcox_data$compared_group != -1, ]
wilcox_data <- melt(wilcox_data, id = c("compared_group"))
wilcox_data <- merge(x = wilcox_data, y = merge(x = var_imp, y = wilcox_res, by.x = "row.names", by.y = "variable"), by.x = "variable", by.y = "Row.names", all.y = TRUE)
wilcox_data$tile_pos <- rep(0, nrow(wilcox_data))
wilcox_data$compared_group <- factor(wilcox_data$compared_group)
wilcox_data$variable <- sapply(wilcox_data$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
adj_pval_tmp <- c()
for (i in 1:nrow(wilcox_data)) {
x <- wilcox_data$adj_pval[i]
if (x >= 1e-3) {
res <- paste0("italic(p) == ", round(x, 3))
} else {
a <- strsplit(format(x, scientific = TRUE, digits = 2), "e")[[1]]
res <- paste0("italic(p) == ", as.numeric(a[1]), " %*% 10^", as.integer(a[2]))
}
adj_pval_tmp <- c(adj_pval_tmp, res)
}
wilcox_data$adj_pval <- adj_pval_tmp
p2 <- ggplot(wilcox_data, aes(x = reorder(variable, `Mean decrease accuracy`), y = value, fill = compared_group)) +
geom_boxplot(outlier.color = NA, alpha = 0.3, lwd = 0.3) +
geom_point(
color = "black", shape = 21, stroke = 0.3, alpha = 0.5, size = 0.5,
position = position_jitterdodge()
) +
geom_text(aes(label = adj_pval), y = 1.1, size = 3, fontface = "plain", family = "sans", hjust = 0, parse = TRUE) +
geom_vline(xintercept = 1:9 + 0.5, color = "grey80", linetype = "solid", size = 0.1) +
scale_x_discrete(
breaks = sapply(c(
"max_parker_hydrophilicity",
"nonsynonymous_snps",
"max_karplus_schulz_flexibility",
"cytoskeletal",
"mitochondrial",
"hydrophobicity_Group3",
"min_score_ifn_epitopes",
"PmN",
"total_snps",
"avg_parker_hydrophilicity"
), function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
}),
labels = c(
"Maximum score of Parker hydrophilicity",
"Number non-synonymous SNPs",
"Maximum score of Karplus and Schulz flexibility",
"Cytoskeletal",
"Mitochondrial",
"% hydrophobicity amino acids",
"Minimum score of IFN-gamma inducing epitopes",
expression("% positive charge a.a. - % negative charge a.a."),
"Total number of SNPs",
"Average score of Parker hydrophilicity"
)
) +
coord_flip(ylim = c(0, 1), clip = "off") +
scale_fill_manual(
breaks = c("1", "0"), values = c("red", "blue"),
labels = c("Candidate antigens", "Random predicted non-antigens")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
strip.background = element_blank(),
panel.border = element_rect(colour = "black"),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(10, 70, 5, 0, "pt"),
legend.text = element_text(colour = "black"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.position = "none"
) +
xlab("") +
ylab("Normalized variable value") +
ggtitle("Group 2 (83 candidates)")wilcox_res <- read.csv("./other_data/candidate_antigen_wilcox_res_3.csv")
wilcox_data <- data
wilcox_data$compared_group <- compared_group_3
var_imp <- read.csv("./other_data/processed_candidate_variable_importance_3.csv", row.names = 1, check.names = FALSE)
wilcox_data <- wilcox_data[wilcox_data$compared_group != -1, ]
wilcox_data <- melt(wilcox_data, id = c("compared_group"))
wilcox_data <- merge(x = wilcox_data, y = merge(x = var_imp, y = wilcox_res, by.x = "row.names", by.y = "variable"), by.x = "variable", by.y = "Row.names", all.y = TRUE)
wilcox_data$tile_pos <- rep(0, nrow(wilcox_data))
wilcox_data$compared_group <- factor(wilcox_data$compared_group)
wilcox_data$variable <- sapply(wilcox_data$variable, function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
})
adj_pval_tmp <- c()
for (i in 1:nrow(wilcox_data)) {
x <- wilcox_data$adj_pval[i]
if (x >= 1e-3) {
res <- paste0("italic(p) == ", round(x, 3))
} else {
a <- strsplit(format(x, scientific = TRUE, digits = 2), "e")[[1]]
res <- paste0("italic(p) == ", as.numeric(a[1]), " %*% 10^", as.integer(a[2]))
}
adj_pval_tmp <- c(adj_pval_tmp, res)
}
wilcox_data$adj_pval <- adj_pval_tmp
p3 <- ggplot(wilcox_data, aes(x = reorder(variable, `Mean decrease accuracy`), y = value, fill = compared_group)) +
geom_boxplot(outlier.color = NA, alpha = 0.3, lwd = 0.3) +
geom_point(
color = "black", shape = 21, stroke = 0.3, alpha = 0.5, size = 0.5,
position = position_jitterdodge()
) +
geom_text(aes(label = adj_pval), y = 1.1, size = 3, fontface = "plain", family = "sans", hjust = 0, parse = TRUE) +
geom_vline(xintercept = 1:9 + 0.5, color = "grey80", linetype = "solid", size = 0.1) +
scale_x_discrete(
breaks = sapply(c(
"out_number_b_cell_epitopes_bepipred_1_0",
"max_parker_hydrophilicity",
"max_karplus_schulz_flexibility",
"nonsynonymous_snps",
"hydrophobicity_Group3",
"cytoskeletal",
"mitochondrial",
"PmN",
"avg_parker_hydrophilicity",
"min_parker_hydrophilicity"
), function(x) {
x <- str_replace_all(x, "[_\\.]", " ")
x <- firstup(x)
return(x)
}),
labels = c(
"Number B-cell epitopes in outer membrane regions",
"Maximum score of Parker hydrophilicity",
"Maximum score of Karplus and Schulz flexibility",
"Number non-synonymous SNPs",
expression("% hydrophobicity amino acids"),
"Cytoskeletal",
"Mitochondrial",
expression("% positive charge a.a. - % negative charge a.a."),
"Average score of Parker hydrophilicity",
"Minimum score of Parker hydrophilicity"
)
) +
coord_flip(ylim = c(0, 1), clip = "off") +
scale_fill_manual(
breaks = c("1", "0"), values = c("red", "blue"),
labels = c("Candidate antigens", "Random predicted non-antigens")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
strip.background = element_blank(),
panel.border = element_rect(colour = "black"),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(10, 70, 5, 20, "pt"),
legend.text = element_text(colour = "black"),
axis.title.x = element_text(color = "black"),
axis.title.y = element_text(color = "black"),
axis.text.x = element_text(color = "black"),
axis.text.y = element_text(color = "black"),
legend.position = "none"
) +
xlab("") +
ylab("Normalized variable value") +
ylim(0, 1) +
ggtitle("Group 3 (56 candidates)")Final plot
p_combined <- plot_grid(plot_grid(p1, p3, ncol = 1, rel_heights = c(0.5, 0.5)),
plot_grid(p2,
NULL,
plot_grid(NULL, legend, nrow = 1, rel_widths = c(0.45, 0.6)),
NULL,
ncol = 1, rel_heights = c(0.5, 0.02, 0.1, 0.4)
),
nrow = 1, rel_widths = c(0.52, 0.48)
)
p_combined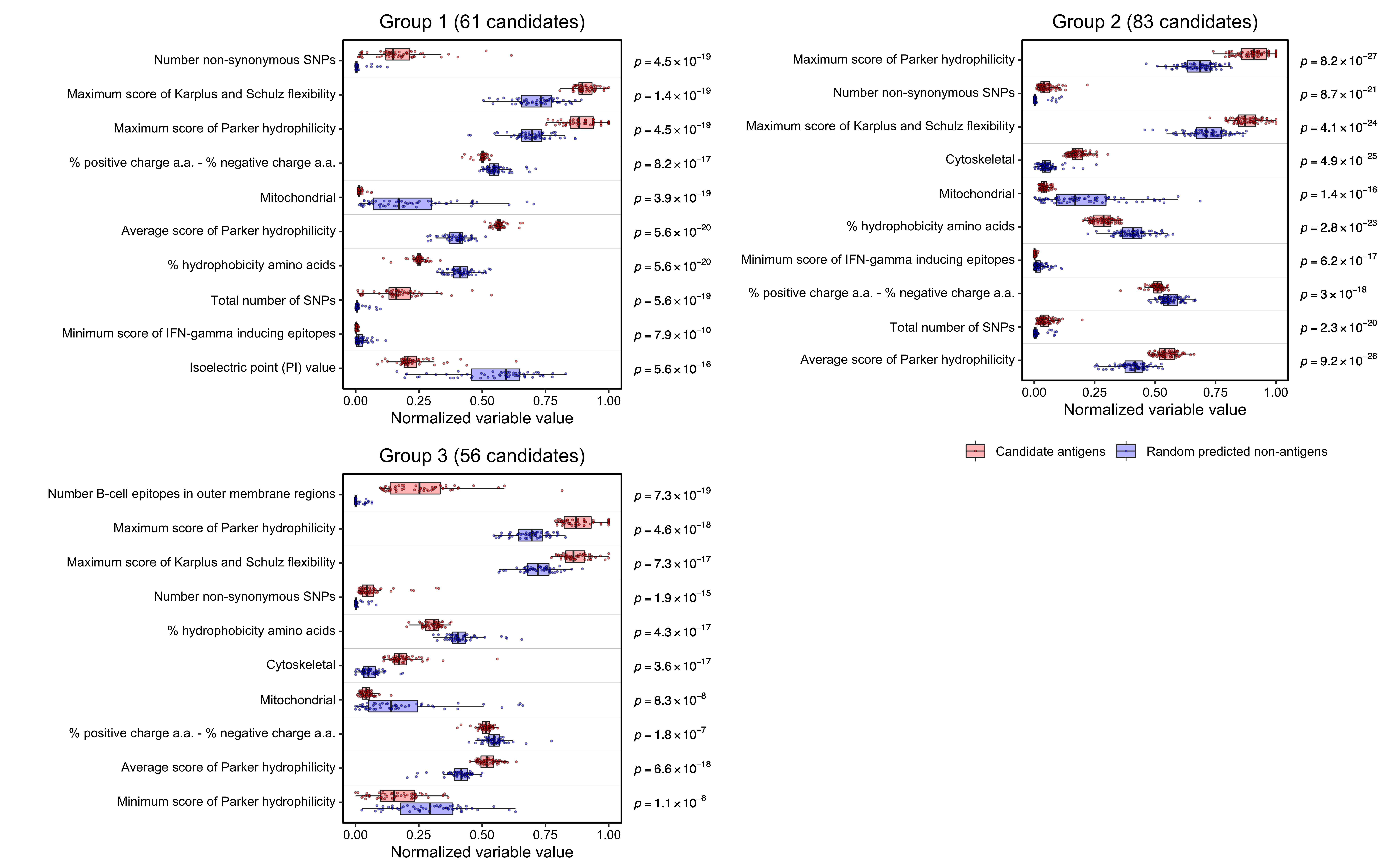
4.4 Comparison of important variables
In R:
var_imp_1 <- read.csv("./other_data/candidate_variable_importance_1.csv", row.names = 1)
var_imp_2 <- read.csv("./other_data/candidate_variable_importance_2.csv", row.names = 1)
var_imp_3 <- read.csv("./other_data/candidate_variable_importance_3.csv", row.names = 1)
var_imp_1 <- var_imp_1[order(-var_imp_1$meanDecreaseAccuracy), , drop = FALSE]
var_imp_2 <- var_imp_2[order(-var_imp_2$meanDecreaseAccuracy), , drop = FALSE]
var_imp_3 <- var_imp_3[order(-var_imp_3$meanDecreaseAccuracy), , drop = FALSE]
var_imp_1$rank_1 <- rank(-var_imp_1$meanDecreaseAccuracy)
var_imp_2$rank_2 <- rank(-var_imp_2$meanDecreaseAccuracy)
var_imp_3$rank_3 <- rank(-var_imp_3$meanDecreaseAccuracy)
var_imp_1$rank_2 <- var_imp_2[rownames(var_imp_1), "rank_2"]
var_imp_1$rank_3 <- var_imp_3[rownames(var_imp_1), "rank_3"]
var_imp_2$rank_1 <- var_imp_1[rownames(var_imp_2), "rank_1"]
var_imp_2$rank_3 <- var_imp_3[rownames(var_imp_2), "rank_3"]
var_imp_3$rank_1 <- var_imp_1[rownames(var_imp_3), "rank_1"]
var_imp_3$rank_2 <- var_imp_2[rownames(var_imp_3), "rank_2"]grp_var_imp_1 <- read.csv("./other_data/candidate_group_variable_importance_1.csv", row.names = 1)
grp_var_imp_2 <- read.csv("./other_data/candidate_group_variable_importance_2.csv", row.names = 1)
grp_var_imp_3 <- read.csv("./other_data/candidate_group_variable_importance_3.csv", row.names = 1)
grp_var_imp_1 <- grp_var_imp_1[order(-grp_var_imp_1$meanDecreaseAccuracy), , drop = FALSE]
grp_var_imp_2 <- grp_var_imp_2[order(-grp_var_imp_2$meanDecreaseAccuracy), , drop = FALSE]
grp_var_imp_3 <- grp_var_imp_3[order(-grp_var_imp_3$meanDecreaseAccuracy), , drop = FALSE]
grp_var_imp_1$rank_1 <- rank(-grp_var_imp_1$meanDecreaseAccuracy)
grp_var_imp_2$rank_2 <- rank(-grp_var_imp_2$meanDecreaseAccuracy)
grp_var_imp_3$rank_3 <- rank(-grp_var_imp_3$meanDecreaseAccuracy)
grp_var_imp_1$rank_2 <- grp_var_imp_2[rownames(grp_var_imp_1), "rank_2"]
grp_var_imp_1$rank_3 <- grp_var_imp_3[rownames(grp_var_imp_1), "rank_3"]
grp_var_imp_2$rank_1 <- grp_var_imp_1[rownames(grp_var_imp_2), "rank_1"]
grp_var_imp_2$rank_3 <- grp_var_imp_3[rownames(grp_var_imp_2), "rank_3"]
grp_var_imp_3$rank_1 <- grp_var_imp_1[rownames(grp_var_imp_3), "rank_1"]
grp_var_imp_3$rank_2 <- grp_var_imp_2[rownames(grp_var_imp_3), "rank_2"]var_imp_1_ <- rbind(var_imp_1[1:10, c("meanDecreaseAccuracy", "rank_2", "rank_3")], grp_var_imp_1[, c("meanDecreaseAccuracy", "rank_2", "rank_3")])
colnames(var_imp_1_) <- c("Mean decrease accuracy", "Group 2 rank", "Group 3 rank")
write.csv(var_imp_1_, "./other_data/processed_candidate_variable_importance_1.csv", row.names = TRUE)
var_imp_2_ <- rbind(var_imp_2[1:10, c("meanDecreaseAccuracy", "rank_1", "rank_3")], grp_var_imp_2[, c("meanDecreaseAccuracy", "rank_1", "rank_3")])
colnames(var_imp_2_) <- c("Mean decrease accuracy", "Group 1 rank", "Group 3 rank")
write.csv(var_imp_2_, "./other_data/processed_candidate_variable_importance_2.csv", row.names = TRUE)
var_imp_3_ <- rbind(var_imp_3[1:10, c("meanDecreaseAccuracy", "rank_1", "rank_2")], grp_var_imp_3[, c("meanDecreaseAccuracy", "rank_1", "rank_2")])
colnames(var_imp_3_) <- c("Mean decrease accuracy", "Group 1 rank", "Group 2 rank")
write.csv(var_imp_3_, "./other_data/processed_candidate_variable_importance_3.csv", row.names = TRUE)Group 1

Group 2

Group 3

sessionInfo()## R version 4.2.3 (2023-03-15)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] sfsmisc_1.1-13 umap_0.2.8.0 cluster_2.1.4 ggforce_0.3.4
## [5] ggbeeswarm_0.6.0 ggpubr_0.4.0 rlist_0.4.6.2 reshape2_1.4.4
## [9] cowplot_1.1.1 ggrepel_0.9.1 stringr_1.4.1 NbClust_3.0.1
## [13] factoextra_1.0.7 ggplot2_3.4.2
##
## loaded via a namespace (and not attached):
## [1] sass_0.4.2 tidyr_1.2.0 jsonlite_1.8.0 carData_3.0-5
## [5] R.utils_2.12.0 bslib_0.4.0 assertthat_0.2.1 askpass_1.1
## [9] highr_0.9 vipor_0.4.5 yaml_2.3.5 pillar_1.8.1
## [13] backports_1.4.1 lattice_0.20-45 glue_1.6.2 reticulate_1.25
## [17] digest_0.6.29 ggsignif_0.6.3 polyclip_1.10-0 colorspace_2.0-3
## [21] htmltools_0.5.3 Matrix_1.5-3 R.oo_1.25.0 plyr_1.8.7
## [25] pkgconfig_2.0.3 broom_1.0.0 bookdown_0.28 purrr_0.3.4
## [29] scales_1.2.1 RSpectra_0.16-1 tweenr_2.0.1 openssl_2.0.2
## [33] tibble_3.1.8 styler_1.8.0 generics_0.1.3 farver_2.1.1
## [37] car_3.1-0 cachem_1.0.6 withr_2.5.0 cli_3.6.1
## [41] magrittr_2.0.3 evaluate_0.16 R.methodsS3_1.8.2 fansi_1.0.3
## [45] R.cache_0.16.0 MASS_7.3-58.2 rstatix_0.7.0 beeswarm_0.4.0
## [49] tools_4.2.3 data.table_1.14.2 lifecycle_1.0.3 munsell_0.5.0
## [53] compiler_4.2.3 jquerylib_0.1.4 rlang_1.1.0 grid_4.2.3
## [57] rstudioapi_0.14 rmarkdown_2.16 codetools_0.2-19 gtable_0.3.0
## [61] abind_1.4-5 DBI_1.1.3 R6_2.5.1 knitr_1.40
## [65] dplyr_1.0.9 fastmap_1.1.0 utf8_1.2.2 stringi_1.7.8
## [69] Rcpp_1.0.9 vctrs_0.6.2 png_0.1-7 tidyselect_1.1.2
## [73] xfun_0.32