Section 2 Model training
2.1 Hyper-parameter tuning for PURF
2.1.1 Analysis
In Python:
library(reticulate)
use_condaenv("/Users/renee/Library/r-miniconda/envs/purf/bin/python")import pandas as pd
from sklearn.utils import shuffle
import numpy as np
import pickle
from sklearn.impute import SimpleImputer
from purf.pu_ensemble import PURandomForestClassifier
from sklearn.preprocessing import MinMaxScaler
from scipy.spatial import distance
from joblib import Parallel, delayed
from sklearn.ensemble._forest import _generate_unsampled_indices
import os
import re
import session_info# private function for train_purf()
def _get_ref_antigen_stats(idx, tree, X, y, ref_indices, max_samples=None):
if max_samples is None:
max_samples = y.shape[0]
oob_indices = _generate_unsampled_indices(tree.random_state, y.shape[0], max_samples)
ref_oob = [i in oob_indices for i in ref_indices]
ref_pred = list()
pred = tree.predict_proba(X[ref_indices,:], check_input=False)
ref_pred = pred[:,1]
return ref_oob, ref_pred
def train_purf(features, outcome, pos_level=0.5, purf=None, tree_filtering=False, ref_antigens=None, n_jobs=1):
features, outcome = shuffle(features, outcome, random_state=0)
# Imputation
imputer = SimpleImputer(strategy='median')
X = imputer.fit_transform(features)
X = pd.DataFrame(X, index=features.index, columns=features.columns)
y = outcome
features = X
print('There are %d positives out of %d samples before feature space weighting.' % (sum(y), len(y)))
# Feature space weighting
lab_pos = X.loc[y==1,:]
median = np.median(lab_pos, axis=0)
# Feature space weighting
lab_pos = X.loc[y==1,:]
median = np.median(lab_pos, axis=0)
scaler = MinMaxScaler(feature_range=(1,10))
dist = list()
for i in range(lab_pos.shape[0]):
dist.append(distance.euclidean(lab_pos.iloc[i, :], median))
dist = np.asarray(dist).reshape(-1, 1)
counts = np.round(scaler.fit_transform(dist))
counts = np.array(counts, dtype=np.int64)[:, 0]
X_temp = X.iloc[y==1, :]
X = X.iloc[y==0, :]
y = np.asarray([0] * X.shape[0] + [1] * (sum(counts)))
appended_data = [X]
for i in range(len(counts)):
appended_data.append(pd.concat([X_temp.iloc[[i]]] * counts[i]))
X = pd.concat(appended_data)
print('There are %d positives out of %d samples after feature space weighting.' % (sum(y), len(y)))
res = pd.DataFrame({'protein_id': X.index, 'antigen_label' : y})
if tree_filtering is True:
# get ref antigen indices
ref_index_dict = {ref:list() for ref in list(ref_antigens.values())}
for i in range(res.shape[0]):
if res['protein_id'][i] in list(ref_antigens.values()):
ref_index_dict[res['protein_id'][i]].append(res.index[i])
ref_indices = sum(ref_index_dict.values(), [])
# get OOB stats and predictions
X = X.astype('float32')
trees = purf.estimators_
idx_list = [i for i in range(len(trees))]
stats_res = Parallel(n_jobs=n_jobs)(
delayed(_get_ref_antigen_stats)(idx, trees[idx], np.array(X), y, ref_indices) for idx in idx_list)
# ref_oob data structure:
# rows represent individual trees
# column represent reference antigens
# cells indicate whether the reference antigen is in the OOB samples of the tree
ref_oob = np.array([ref_oob for ref_oob, ref_pred in stats_res])
# ref_pred data structure:
# rows represent individual trees
# column represent reference antigens
# cells indicate the prediction of the reference antigen by the tree
ref_pred = np.array([ref_pred for ref_oob, ref_pred in stats_res])
# analyze duplicated reference antigens as a group
cumsum_num_ref = np.cumsum(np.array([len(v) for k,v in ref_index_dict.items()]))
ref_oob_all = np.array([ref_oob[:, 0:cumsum_num_ref[i]].any(axis=1) if i == 0 else \
ref_oob[:, cumsum_num_ref[i - 1]:cumsum_num_ref[i]].any(axis=1) \
for i in range(len(ref_antigens))]).T
ref_pred_all = np.array([ref_pred[:, 0:cumsum_num_ref[i]].any(axis=1) if i == 0 else \
ref_pred[:, cumsum_num_ref[i - 1]:cumsum_num_ref[i]].sum(axis=1) \
for i in range(len(ref_antigens))]).T
# calculate number of reference antigens as OOB samples for each tree
oob_total = ref_oob_all.sum(axis=1)
# assign score of 1 to trees that correctly predict all OOB reference antigens; otherwise, assign 0 score
weights = np.zeros(len(trees))
# iterate through the trees and calculate the stats
for i in range(len(trees)):
oob_list = list(ref_oob_all[i,:])
pred_list = list(ref_pred_all[i,:])
if oob_total[i] == 0:
weights[i] = 0
else:
if sum(np.array(pred_list)[oob_list] != 0) == oob_total[i]:
weights[i] = 1
if tree_filtering is False:
# Training PURF
purf = PURandomForestClassifier(
n_estimators = 100000,
oob_score = True,
n_jobs = 64,
random_state = 42,
pos_level = pos_level
)
purf.fit(X, y)
else:
purf._set_oob_score_with_weights(np.array(X), y.reshape(-1,1), weights=weights)
# Storing results
res['OOB score'] = purf.oob_decision_function_[:,1]
res = features.merge(res.groupby('protein_id').mean(), left_index=True, right_on='protein_id')
res = res[['antigen_label', 'OOB score']]
if tree_filtering is False:
return (purf, res)
else:
return ({'model': purf, 'weights': weights}, res)2.1.1.1 Pv data set
ref_antigens = {'CSP': 'PVP01_0835600.1-p1', 'DBP': 'PVP01_0623800.1-p1', 'MSP1': 'PVP01_0728900.1-p1'}
data = pd.read_csv('./other_data/pv_ml_input.csv', index_col=0)
features = data.iloc[:, 1:]
outcome = np.array(data.antigen_label)
for pos_level in [0.5, 0.4, 0.6, 0.3, 0.7, 0.2, 0.8, 0.1, 0.9]:
(purf, res) = train_purf(features, outcome, pos_level=pos_level, tree_filtering=False)
(purf_filtered, res_filtered) = train_purf(features, outcome, pos_level=pos_level, purf=purf, tree_filtering=True, ref_antigens=ref_antigens)
res['OOB score filtered'] = res_filtered['OOB score']
res.to_csv('~/Downloads/pv_pos_level/%.1f_res.tsv' % pos_level)
with open('~/Downloads/pv_pos_level/%.1f_purf_tree_filtering.pkl' % pos_level, 'wb') as out:
pickle.dump(purf_filtered, out, pickle.HIGHEST_PROTOCOL)dir = '~/Downloads/pv_pos_level/'
files = os.listdir(dir)
tmp = pd.read_csv(dir + '0.1_res.tsv', sep='\t', index_col=0)['antigen_label']
data_frames = [pd.read_csv(dir + '%.1f_res.tsv' % pos_level, sep='\t', index_col=0)['OOB score filtered'] for pos_level in np.arange(0.1, 1, 0.1)]
merged_df = pd.concat([tmp] + data_frames, join='outer', axis=1)
colnames = ['antigen_label'] + ['%.1f' % pos_level for pos_level in np.arange(0.1, 1, 0.1)]
merged_df.columns = colnames
merged_df.to_csv('./other_data/pv_pos_level_parameter_tuning.csv')2.1.1.2 Pv + Pf combined data set
ref_antigens = {'CSP (Pf)': 'PF3D7_0304600.1-p1', 'RH5 (Pf)': 'PF3D7_0424100.1-p1', 'MSP5 (Pf)': 'PF3D7_0206900.1-p1',
'P230 (Pf)': 'PF3D7_0209000.1-p1',
'CSP (Pv)': 'PVP01_0835600.1-p1', 'DBP (Pv)': 'PVP01_0623800.1-p1', 'MSP1 (Pv)': 'PVP01_0728900.1-p1'}
data = pd.read_csv('./data/supplementary_data_4_pfpv_ml_input.csv', index_col=0)
features = data.iloc[:, 1:]
outcome = np.array(data.antigen_label)
for pos_level in [0.5, 0.4, 0.6, 0.3, 0.7, 0.2, 0.8, 0.1, 0.9]:
(purf, res) = train_purf(features, outcome, pos_level=pos_level, tree_filtering=False)
(purf_filtered, res_filtered) = train_purf(features, outcome, pos_level=pos_level, purf=purf, tree_filtering=True, ref_antigens=ref_antigens)
res['OOB score filtered'] = res_filtered['OOB score']
res.to_csv('~/Downloads/pfpv_pos_level/%.1f_res.tsv' % pos_level)
with open('~/Downloads/pfpv_pos_level/%.1f_purf_tree_filtering.pkl' % pos_level, 'wb') as out:
pickle.dump(purf_filtered, out, pickle.HIGHEST_PROTOCOL)dir = '~/Downloads/pfpv_pos_level/'
files = os.listdir(dir)
tmp = pd.read_csv(dir + '0.1_res.tsv', sep='\t', index_col=0)['antigen_label']
data_frames = [pd.read_csv(dir + '%.1f_res.tsv' % pos_level, sep='\t', index_col=0)['OOB score filtered'] for pos_level in np.arange(0.1, 1, 0.1)]
merged_df = pd.concat([tmp] + data_frames, join='outer', axis=1)
colnames = ['antigen_label'] + ['%.1f' % pos_level for pos_level in np.arange(0.1, 1, 0.1)]
merged_df.columns = colnames
merged_df.to_csv('./other_data/pfpv_pos_level_parameter_tuning.csv')2.1.2 Plotting
In R:
library(mixR)
library(pracma)
library(rlist)
library(ggplot2)
library(cowplot)
library(grid)
library(gridExtra)2.1.2.1 Pv data set
data <- read.csv("./other_data/pv_pos_level_parameter_tuning.csv", header = TRUE, row.names = 1, check.names = FALSE)
# Extract data with only unlabeled proteins
data_unl <- data[data$antigen_label == 0, ]plot_list <- list()
plot_list2 <- list()
for (i in seq(0.1, 0.9, 0.1)) {
fit <- mixfit(data_unl[[as.character(i)]], ncomp = 2)
# Calculate receiver operating characteristic (ROC) curve
# for putative positive and negative samples
x <- seq(-0.5, 1.5, by = 0.01)
neg_cum <- pnorm(x, mean = fit$mu[1], sd = fit$sd[1])
pos_cum <- pnorm(x, mean = fit$mu[2], sd = fit$sd[2])
fpr <- (1 - neg_cum) / ((1 - neg_cum) + neg_cum) # false positive / (false positive + true negative)
tpr <- (1 - pos_cum) / ((1 - pos_cum) + pos_cum) # true positive / (true positive + false negative)
p <- plot(fit, title = paste0(
"Positive level = ", i,
" (AUROC = ", round(trapz(-fpr, tpr), 2), ")"
)) +
scale_fill_manual(values = c("#0080FF", "#FF007F"), labels = c("Putative negative", "Putative positive")) +
theme_bw() +
{
if (i == 0.1) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = c(0.7, 0.85),
legend.background = element_blank()
)
} else {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
)
}
} +
xlim(-0.8, 1.5)
# Calculate percent rank for labeled positives
data_ <- data[c("antigen_label", as.character(i))]
colnames(data_) <- c("antigen_label", "OOB score")
data_$percent_rank <- rank(data_[["OOB score"]]) / nrow(data)
data_ <- data_[data$antigen_label == 1, ]
data_ <- data_[order(-data_$percent_rank), ]
data_$x <- 1:nrow(data_) / nrow(data_)
cat(paste0("EPR: ", sum(data_$`OOB score` >= 0.5) / nrow(data_), "\n"))
p2 <- ggplot(data_, aes(x = x, y = `percent_rank`)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "grey50") +
geom_line(linewidth = 0.1, color = "grey30") +
geom_point(aes(fill = `OOB score`), size = 2.2, shape = 21, color = "grey30", stroke = 0.1) +
scale_fill_gradient2(low = "#0080FF", mid = "white", high = "#FF007F", midpoint = 0.5, limits = c(0, 1)) +
theme_bw() +
{
if (i == 0.1) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.5, 0.15),
legend.background = element_blank(),
legend.title.align = -130
)
} else {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
)
}
} +
{
if (i == 0.1) {
guides(fill = guide_colourbar(title.position = "top", direction = "horizontal"))
}
} +
{
if (i == 0.1) {
labs(fill = "Score (proportion of votes)")
}
} +
ggtitle(paste0(
"Positive level = ", i,
" (AUC = ", round(trapz(c(0, data_$x, 1), c(1, data_$percent_rank, 0)), 2), ")"
)) +
ylim(0, 1)
plot_list <- list.append(plot_list, p)
plot_list2 <- list.append(plot_list2, p2)
}2.1.2.2 Bimodal distribution plot
x_grob <- textGrob("Score (proportion of votes)", gp = gpar(fontsize = 15))
y_grob <- textGrob("Density", gp = gpar(fontsize = 15), rot = 90)
grid.arrange(arrangeGrob(plot_grid(plotlist = plot_list, ncol = 3), left = y_grob, bottom = x_grob))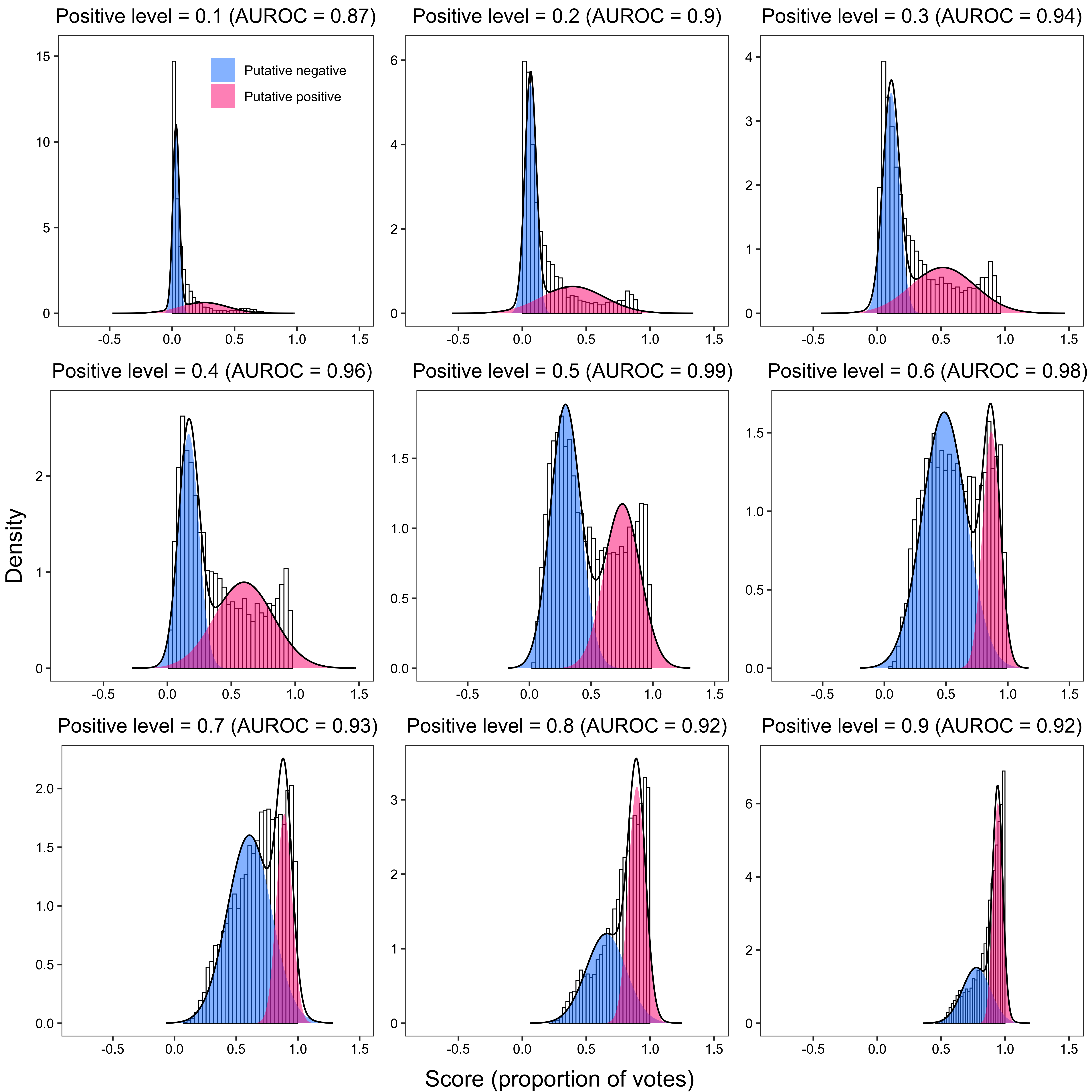
2.1.2.3 Known antigen ranking
x_grob2 <- textGrob("Ranked known antigens (scaled)", gp = gpar(fontsize = 15))
y_grob2 <- textGrob("Percent rank", gp = gpar(fontsize = 15), rot = 90)
grid.arrange(arrangeGrob(plot_grid(plotlist = plot_list2, ncol = 3), left = y_grob2, bottom = x_grob2))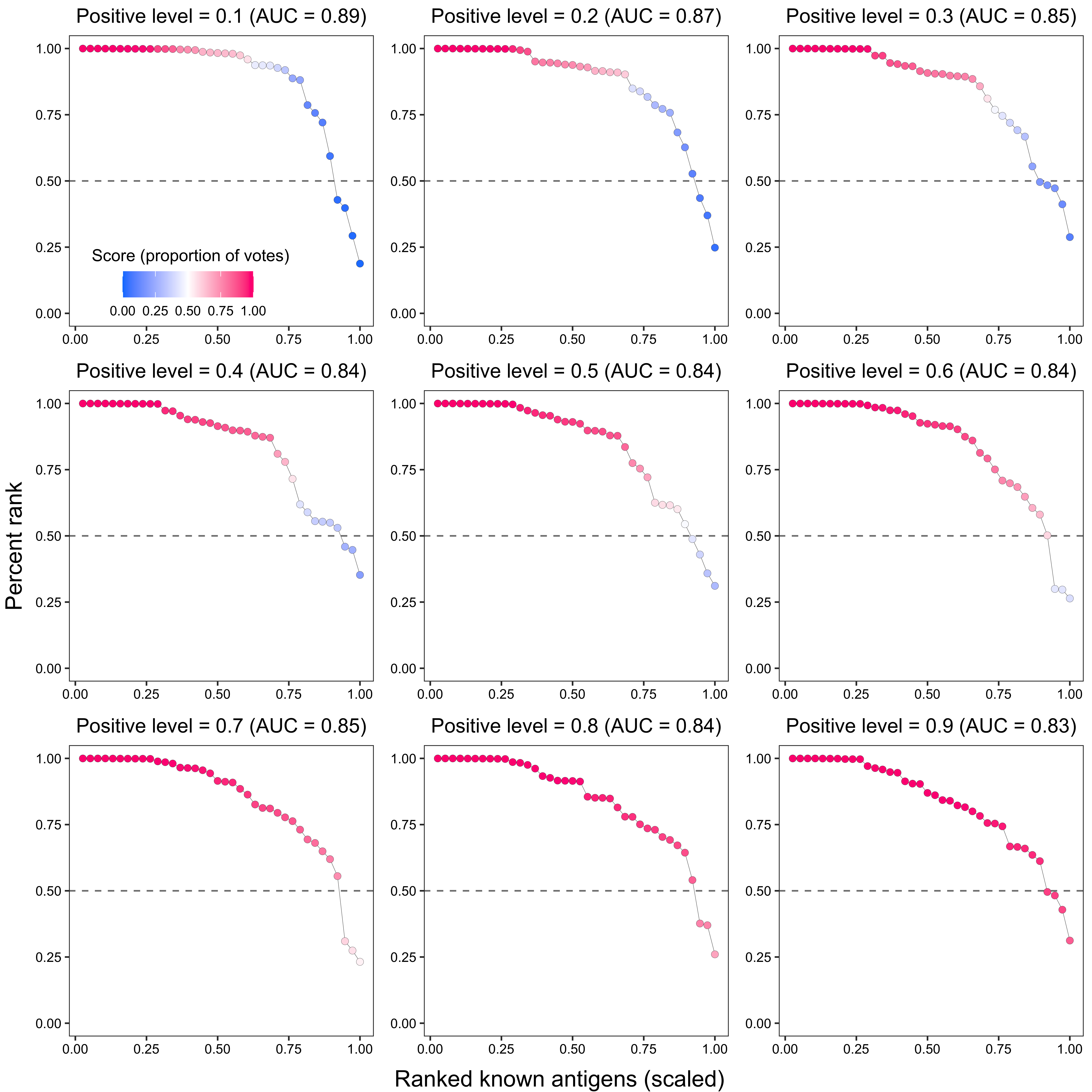
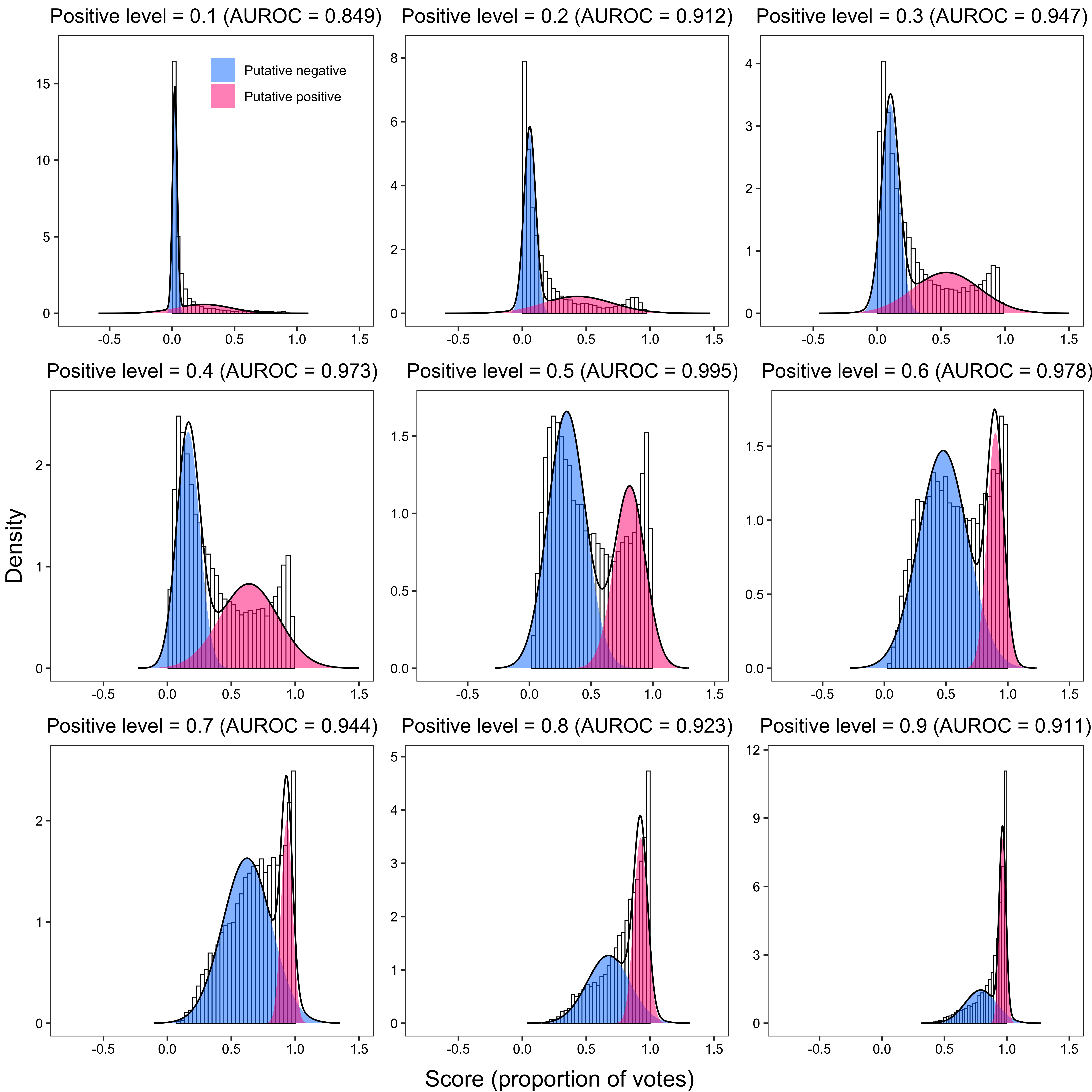
2.1.2.4 Pv + Pf combined data set
data <- read.csv("./other_data/pfpv_pos_level_parameter_tuning.csv", header = TRUE, row.names = 1, check.names = FALSE)
# Extract data with only unlabeled proteins
data_unl <- data[data$antigen_label == 0, ]plot_list3 <- list()
plot_list4 <- list()
for (i in seq(0.1, 0.9, 0.1)) {
fit <- mixfit(data_unl[[as.character(i)]], ncomp = 2)
# Calculate receiver operating characteristic (ROC) curve
# for putative positive and negative samples
x <- seq(-0.5, 1.5, by = 0.01)
neg_cum <- pnorm(x, mean = fit$mu[1], sd = fit$sd[1])
pos_cum <- pnorm(x, mean = fit$mu[2], sd = fit$sd[2])
fpr <- (1 - neg_cum) / ((1 - neg_cum) + neg_cum) # false positive / (false positive + true negative)
tpr <- (1 - pos_cum) / ((1 - pos_cum) + pos_cum) # true positive / (true positive + false negative)
p <- plot(fit, title = paste0(
"Positive level = ", i,
" (AUROC = ", round(trapz(-fpr, tpr), 3), ")"
)) +
scale_fill_manual(values = c("#0080FF", "#FF007F"), labels = c("Putative negative", "Putative positive")) +
theme_bw() +
{
if (i == 0.1) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = c(0.7, 0.85),
legend.background = element_blank()
)
} else {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
)
}
} +
xlim(-0.8, 1.5)
# Calculate percent rank for labeled positives
data_ <- data[c("antigen_label", as.character(i))]
colnames(data_) <- c("antigen_label", "OOB score")
data_$percent_rank <- rank(data_[["OOB score"]]) / nrow(data)
data_ <- data_[data$antigen_label == 1, ]
data_ <- data_[order(-data_$percent_rank), ]
data_$x <- 1:nrow(data_) / nrow(data_)
cat(paste0("EPR: ", sum(data_$`OOB score` >= 0.5) / nrow(data_), "\n"))
p2 <- ggplot(data_, aes(x = x, y = `percent_rank`)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "grey50") +
geom_line(linewidth = 0.1, color = "grey30") +
geom_point(aes(fill = `OOB score`), size = 1.2, shape = 21, color = "grey30", stroke = 0.1) +
scale_fill_gradient2(low = "#0080FF", mid = "white", high = "#FF007F", midpoint = 0.5, limits = c(0, 1)) +
theme_bw() +
{
if (i == 0.1) {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.5, 0.15),
legend.background = element_blank(),
legend.title.align = -130
)
} else {
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.position = "none"
)
}
} +
{
if (i == 0.1) {
guides(fill = guide_colourbar(title.position = "top", direction = "horizontal"))
}
} +
{
if (i == 0.1) {
labs(fill = "Score (proportion of votes)")
}
} +
ggtitle(paste0(
"Positive level = ", i,
" (AUC = ", round(trapz(c(0, data_$x, 1), c(1, data_$percent_rank, 0)), 2), ")"
)) +
ylim(0, 1)
plot_list3 <- list.append(plot_list3, p)
plot_list4 <- list.append(plot_list4, p2)
}
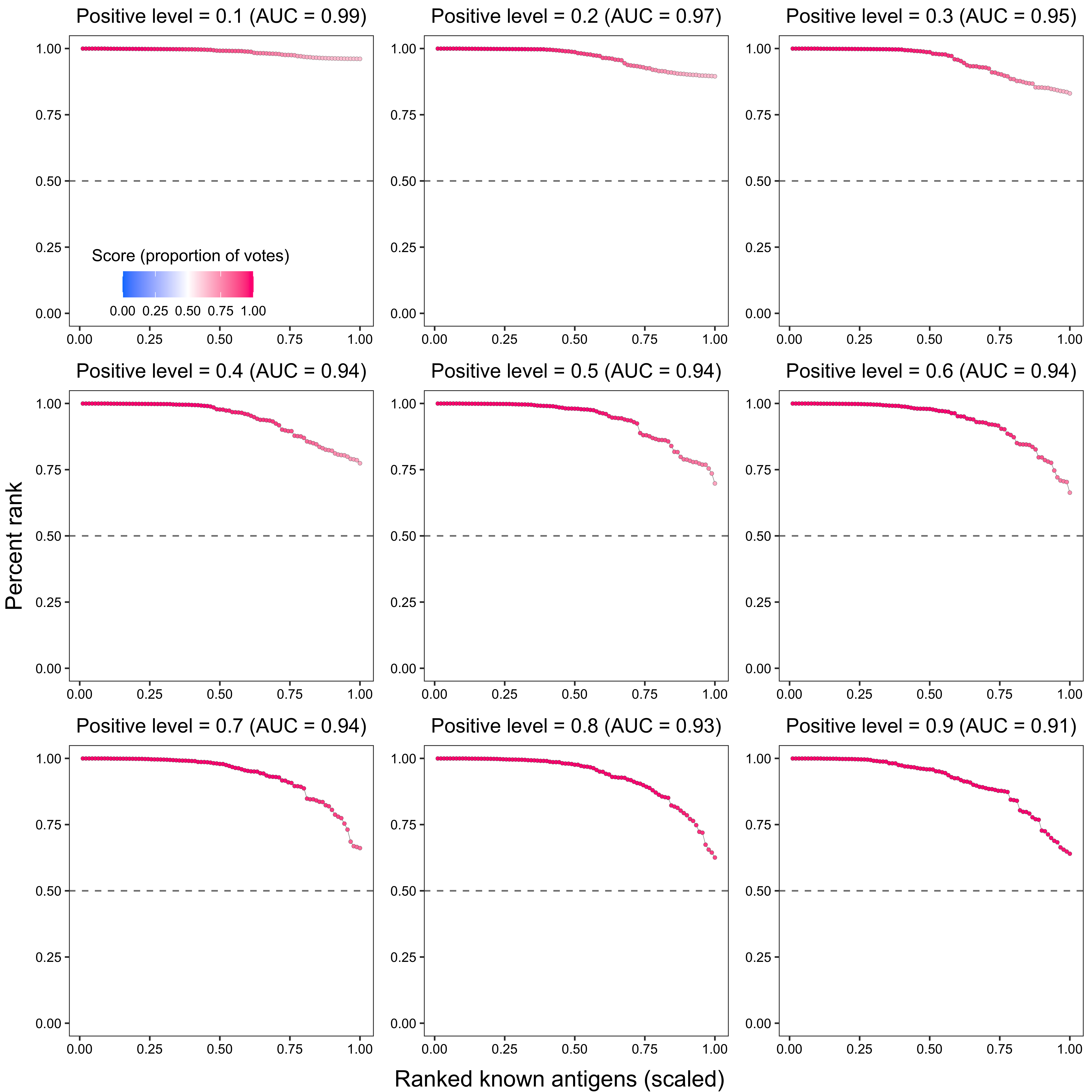
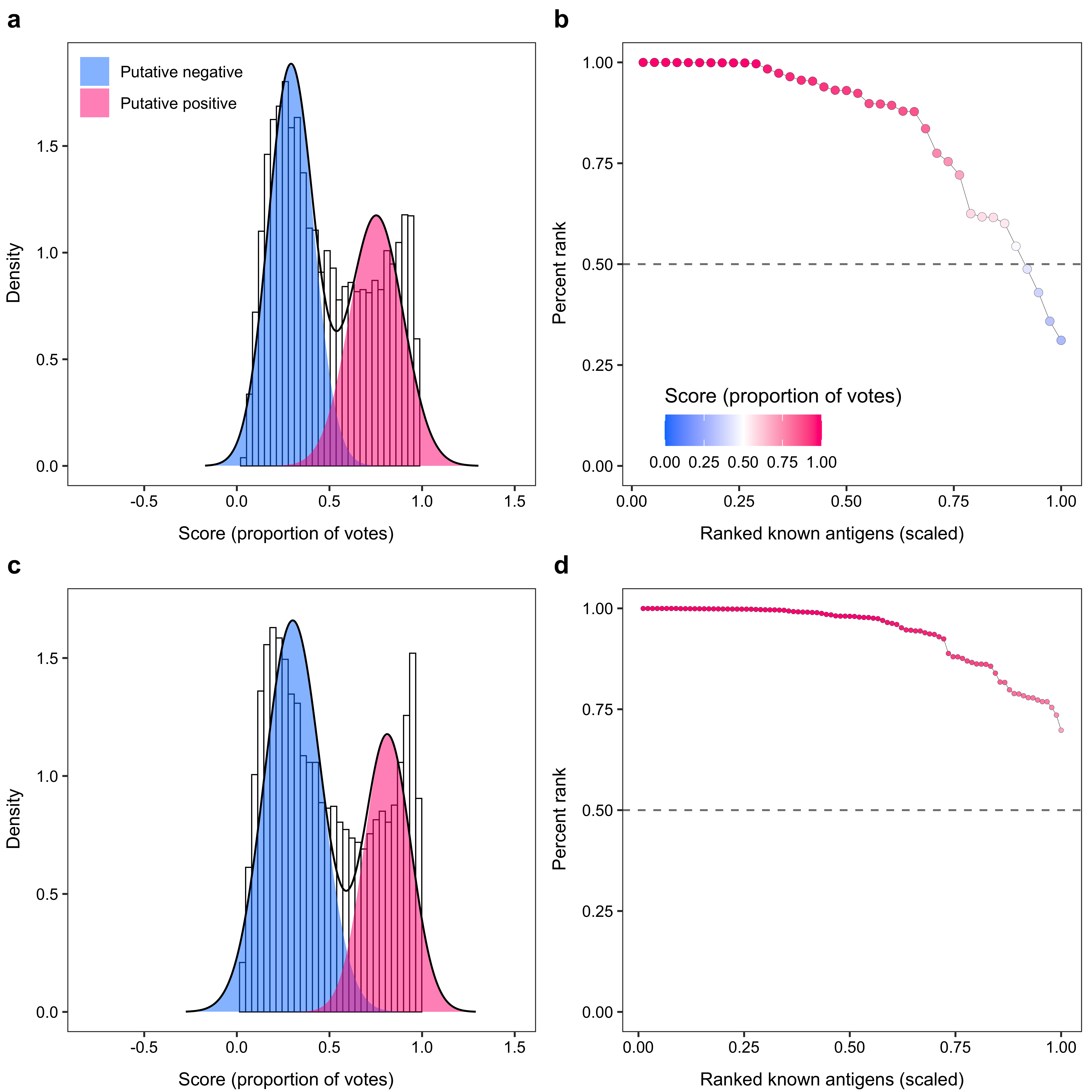
2.1.3 Results for positive level = 0.5
plot_list[[5]]$labels$title <- ""
plot_list2[[5]]$labels$title <- ""
plot_list3[[5]]$labels$title <- ""
plot_list4[[5]]$labels$title <- ""
plot_list[[5]] <- plot_list[[5]] +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = c(0.2, 0.91),
legend.background = element_blank()
)
plot_list2[[5]] <- plot_list2[[5]] +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.35, 0.13),
legend.background = element_blank(),
legend.title.align = -130
) +
guides(fill = guide_colourbar(title.position = "top", direction = "horizontal")) +
labs(fill = "Score (proportion of votes)")
x_grob <- textGrob("Score (proportion of votes)", gp = gpar(fontsize = 10))
y_grob <- textGrob("Density", gp = gpar(fontsize = 10), rot = 90)
x_grob2 <- textGrob("Ranked known antigens (scaled)", gp = gpar(fontsize = 10))
y_grob2 <- textGrob("Percent rank", gp = gpar(fontsize = 10), rot = 90)
plot_grid(
arrangeGrob(plot_list[[5]], left = y_grob, bottom = x_grob),
arrangeGrob(plot_list2[[5]], left = y_grob2, bottom = x_grob2),
arrangeGrob(plot_list3[[5]], left = y_grob, bottom = x_grob),
arrangeGrob(plot_list4[[5]], left = y_grob2, bottom = x_grob2),
nrow = 2, labels = c("a", "b", "c", "d")
)
sessionInfo()## R version 4.2.3 (2023-03-15)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] gridExtra_2.3 cowplot_1.1.1 ggplot2_3.4.2 rlist_0.4.6.2
## [5] pracma_2.4.2 mixR_0.2.0 reticulate_1.28
##
## loaded via a namespace (and not attached):
## [1] styler_1.9.1 tidyselect_1.2.0 xfun_0.39 bslib_0.4.2
## [5] purrr_1.0.1 lattice_0.21-8 colorspace_2.1-0 vctrs_0.6.2
## [9] generics_0.1.3 htmltools_0.5.5 yaml_2.3.7 utf8_1.2.3
## [13] rlang_1.1.1 R.oo_1.25.0 jquerylib_0.1.4 pillar_1.9.0
## [17] glue_1.6.2 withr_2.5.0 R.utils_2.12.2 R.cache_0.16.0
## [21] lifecycle_1.0.3 munsell_0.5.0 gtable_0.3.3 R.methodsS3_1.8.2
## [25] codetools_0.2-19 evaluate_0.21 knitr_1.42 fastmap_1.1.1
## [29] fansi_1.0.4 highr_0.10 Rcpp_1.0.10 scales_1.2.1
## [33] cachem_1.0.8 jsonlite_1.8.4 png_0.1-8 digest_0.6.31
## [37] bookdown_0.34 dplyr_1.1.2 rprojroot_2.0.3 here_1.0.1
## [41] cli_3.6.1 tools_4.2.3 magrittr_2.0.3 sass_0.4.6
## [45] tibble_3.2.1 pkgconfig_2.0.3 Matrix_1.5-4 data.table_1.14.8
## [49] rmarkdown_2.21 rstudioapi_0.14 R6_2.5.1 compiler_4.2.3session_info.show()## -----
## joblib 1.1.1
## numpy 1.19.0
## pandas 1.3.2
## purf NA
## scipy 1.8.0
## session_info 1.0.0
## sklearn 0.24.2
## -----
## Python 3.8.2 (default, Mar 26 2020, 10:45:18) [Clang 4.0.1 (tags/RELEASE_401/final)]
## macOS-10.16-x86_64-i386-64bit
## -----
## Session information updated at 2023-05-18 12:34