Section 4 Model comparisons
4.1 Known antigen score comparisons
In R:
library(ggplot2)
library(reshape2)
library(ggdist)
library(gghalves)
library(cowplot)pv_scores <- read.csv("./other_data/pv_known_antigen_scores.csv", row.names = 1)
pv_unl_scores <- read.csv("./other_data/pv_unlabeled_protein_scores.csv", row.names = 1)
pv_scores$label <- 2
pv_unl_scores$label <- 1
data_ <- melt(rbind(pv_scores, pv_unl_scores), id.vars = "label")
data_$variable <- factor(data_$variable, levels = c(
"pf_single_model",
"pv_single_model", "pv_pf_pos",
"pfpv_combined_model",
"pv_pf_unl"
))
data_$label[data_$value < 0.5 & data_$label == 1] <- 0
set.seed(12)
p1 <- ggplot(data_, aes(x = variable, y = value)) +
geom_hline(yintercept = 0.5, color = "grey70", linetype = "dashed") +
stat_halfeye(aes(fill = factor(label, levels = c(2, 1, 0))),
slab_color = "black",
slab_linewidth = 0.2, adjust = 0.5, width = 0.5, .width = 0,
justification = -0.2, point_colour = NA, alpha = 0.6, normalize = "all"
) +
geom_boxplot(aes(fill = factor(label, levels = c(2, 1, 0))),
width = 0.2, outlier.color = NA,
lwd = 0.3, show.legend = FALSE, alpha = 0.6, color = "black"
) +
geom_half_point(
data = data_[data_$label == 2, ], fill = "#FCB40A", side = "l",
range_scale = 0.3, alpha = 0.8, shape = 21, color = "black", stroke = 0.1, size = 2
) +
scale_fill_manual(
name = "", values = c("#FCB40A", "#FF007F", "#0080FF"), breaks = c(2, 1, 0),
labels = c("Known antigen", "Predicted antigen", "Predicted non-antigen")
) +
scale_color_manual(
name = "", values = c("#FCB40A", "#FF007F", "#0080FF"), breaks = c(2, 1, 0),
labels = c("Known antigen", "Predicted antigen", "Predicted non-antigen")
) +
coord_flip() +
scale_x_discrete(
breaks = c(
"pf_single_model", "pv_single_model", "pv_pf_pos",
"pfpv_combined_model", "pv_pf_unl"
),
labels = c(
"Heterologous model", "Autologous model",
"Autologous with\n heterologous positives model",
"Combined model",
"Autologous with\n heterologous unlabeled model"
),
limits = rev
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title.x = element_text(colour = "black"),
axis.title.y = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
legend.position = "none"
) +
ylim(0, 1) +
ylab("Score (proportion of votes)")
legend <- get_legend(p2 +
theme(
legend.direction = "horizontal",
legend.position = "bottom",
legend.title = element_blank()
) +
guides(fill = guide_legend(title = "")))pf_scores <- read.csv("./other_data/pf_known_antigen_scores.csv", row.names = 1)
pf_unl_scores <- read.csv("./other_data/pf_unlabeled_protein_scores.csv", row.names = 1)
pf_scores$label <- 2
pf_unl_scores$label <- 1
data_ <- melt(rbind(pf_scores, pf_unl_scores), id.vars = "label")
data_$variable <- factor(data_$variable, levels = c(
"pv_single_model",
"pf_single_model", "pf_pv_pos",
"pfpv_combined_model",
"pf_pv_unl"
))
data_$label[data_$value < 0.5 & data_$label == 1] <- 0
set.seed(12)
p2 <- ggplot(data_, aes(x = variable, y = value)) +
geom_hline(yintercept = 0.5, color = "grey70", linetype = "dashed") +
stat_halfeye(aes(fill = factor(label, levels = c(2, 1, 0))),
slab_color = "black",
slab_linewidth = 0.2, adjust = 0.5, width = 0.5, .width = 0,
justification = -0.2, point_colour = NA, alpha = 0.6, normalize = "all"
) +
geom_boxplot(aes(fill = factor(label, levels = c(2, 1, 0))),
width = 0.2, outlier.color = NA,
lwd = 0.3, show.legend = FALSE, alpha = 0.6, color = "black"
) +
geom_half_point(
data = data_[data_$label == 2, ], fill = "#FCB40A", side = "l",
range_scale = 0.3, alpha = 0.8, shape = 21, color = "black", stroke = 0.1, size = 2
) +
scale_fill_manual(
name = "", values = c("#FCB40A", "#FF007F", "#0080FF"), breaks = c(2, 1, 0),
labels = c("Known antigen", "Predicted antigen", "Predicted non-antigen")
) +
scale_color_manual(
name = "", values = c("#FCB40A", "#FF007F", "#0080FF"), breaks = c(2, 1, 0),
labels = c("Known antigen", "Predicted antigen", "Predicted non-antigen")
) +
coord_flip() +
scale_x_discrete(
breaks = c(
"pv_single_model", "pf_single_model", "pf_pv_pos",
"pfpv_combined_model", "pf_pv_unl"
),
labels = c(
"Heterologous model", "Autologous model",
"Autologous with\n heterologous positives model",
"Combined model",
"Autologous with\n heterologous unlabeled model"
),
limits = rev
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title.x = element_text(colour = "black"),
axis.title.y = element_blank(),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
legend.title = element_blank(),
legend.text = element_text(colour = "black"),
legend.position = "none"
) +
ylim(0, 1) +
ylab("Score (proportion of votes)")p_combined <- plot_grid(p1, p2, plot_grid(NULL, legend, rel_widths = c(0.2, 0.8)),
ncol = 2,
rel_heights = c(1, 0.1), labels = c("a", "b", "", "")
)
p_combinedpng(file = "./figures/Fig 2.png", width = 7500, height = 4000, res = 600)
print(p_combined)
dev.off()
pdf(file = "../figures/Fig 2.pdf", width = 13, height = 7)
print(p_combined)
dev.off()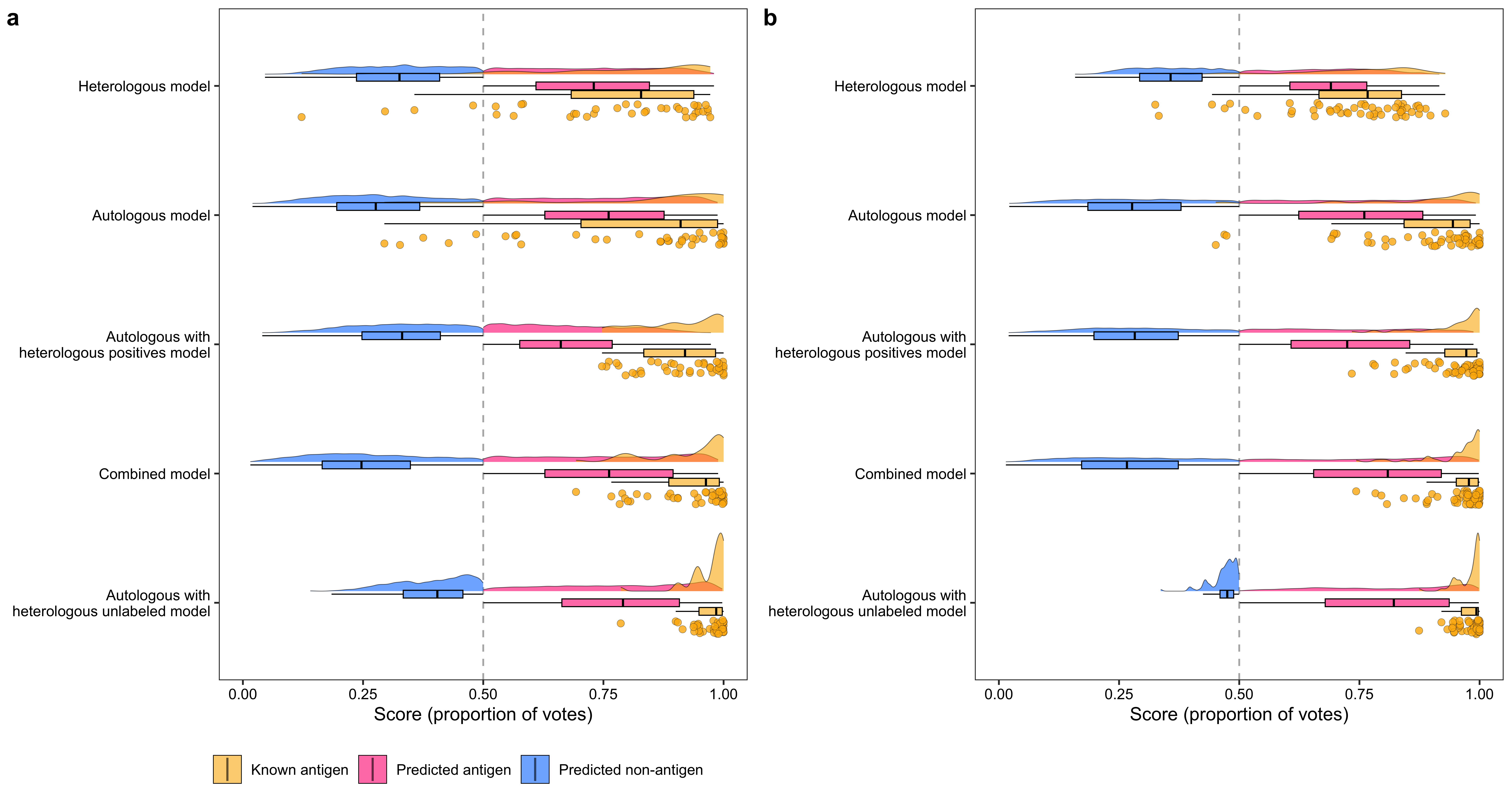
4.2 Known antigen prediction summary
In R:
library(pracma)
library(ggsci)pv_scores <- read.csv("./other_data/pv_known_antigen_scores.csv", row.names = 1)
pv_unl_scores <- read.csv("./other_data/pv_unlabeled_protein_scores.csv", row.names = 1)
pv_scores$label <- 1
pv_unl_scores$label <- 0
pv_all <- rbind(pv_scores, pv_unl_scores)
data <- data.frame(
"antigen_label" = pv_all$label,
"Heterologous model" = pv_all$pf_single_model,
"Autologous model" = pv_all$pv_single_model,
"Autologous with heterologous positives model" = pv_all$pv_pf_pos,
"Combined model" = pv_all$pfpv_combined_model,
"Autologous with heterologous unlabeled model" = pv_all$pv_pf_unl,
check.names = FALSE
)
# Calculate percent rank for labeled positives
model_names <- c()
x <- c()
percent_rank <- c()
auc <- c()
for (model in colnames(data)[-1]) {
data_ <- data[c("antigen_label", model)]
percent_rank_ <- (rank(data_[[model]]) / nrow(data))[data$antigen_label == 1]
percent_rank <- c(percent_rank, percent_rank_[order(-percent_rank_)])
data_ <- data_[data$antigen_label == 1, ]
data_ <- data_[order(-percent_rank_), ]
x_ <- 1:nrow(data_) / nrow(data_)
x <- c(x, x_)
model_names <- c(model_names, rep(model, nrow(data_)))
auc <- c(auc, round(trapz(c(0, x_, 1), c(1, percent_rank_, 0)), 2))
cat(paste0("EPR: ", sum(data_[[model]] >= 0.5) / nrow(data_), "\n"))
}
names(auc) <- colnames(data)[-1]
res <- data.frame(model = model_names, x = x, percent_rank = percent_rank)
p1 <- ggplot(res, aes(x = x, y = `percent_rank`, group = model)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "grey50") +
geom_line(linewidth = 0.1, color = "grey30") +
geom_point(aes(fill = model),
size = 2.2, shape = 21, color = "grey30", stroke = 0.1,
alpha = 0.8
) +
scale_fill_jco(
breaks = colnames(data)[-1][order(-auc)],
labels = sapply(colnames(data)[-1][order(-auc)], function(x) {
paste0(
x, " (AUC = ",
auc[x], ")"
)
})
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.34, 0.16),
legend.background = element_blank(),
legend.key = element_rect(colour = "transparent", fill = "transparent")
) +
labs(fill = "") +
ylim(0, 1) +
xlab("Ranked known antigens (scaled)") +
ylab("Percent rank")pf_scores <- read.csv("./other_data/pf_known_antigen_scores.csv", row.names = 1)
pf_unl_scores <- read.csv("./other_data/pf_unlabeled_protein_scores.csv", row.names = 1)
pf_scores$label <- 1
pf_unl_scores$label <- 0
pf_all <- rbind(pf_scores, pf_unl_scores)
data <- data.frame(
"antigen_label" = pf_all$label,
"Heterologous model" = pf_all$pv_single_model,
"Autologous model" = pf_all$pf_single_model,
"Autologous with heterologous positives model" = pf_all$pf_pv_pos,
"Combined model" = pf_all$pfpv_combined_model,
"Autologous with heterologous unlabeled model" = pf_all$pf_pv_unl,
check.names = FALSE
)
# Calculate percent rank for labeled positives
model_names <- c()
x <- c()
percent_rank <- c()
auc <- c()
for (model in colnames(data)[-1]) {
data_ <- data[c("antigen_label", model)]
percent_rank_ <- (rank(data_[[model]]) / nrow(data))[data$antigen_label == 1]
percent_rank <- c(percent_rank, percent_rank_[order(-percent_rank_)])
data_ <- data_[data$antigen_label == 1, ]
data_ <- data_[order(-percent_rank_), ]
x_ <- 1:nrow(data_) / nrow(data_)
x <- c(x, x_)
model_names <- c(model_names, rep(model, nrow(data_)))
auc <- c(auc, round(trapz(c(0, x_, 1), c(1, percent_rank_, 0)), 2))
cat(paste0("EPR: ", sum(data_[[model]] >= 0.5) / nrow(data_), "\n"))
}
names(auc) <- colnames(data)[-1]
res <- data.frame(model = model_names, x = x, percent_rank = percent_rank)
p2 <- ggplot(res, aes(x = x, y = `percent_rank`, group = model)) +
geom_hline(yintercept = 0.5, linetype = "dashed", color = "grey50") +
geom_line(linewidth = 0.1, color = "grey30") +
geom_point(aes(fill = model),
size = 2.2, shape = 21, color = "grey30", stroke = 0.1,
alpha = 0.8
) +
scale_fill_jco(
breaks = colnames(data)[-1][order(-auc)],
labels = sapply(colnames(data)[-1][order(-auc)], function(x) {
paste0(
x, " (AUC = ",
auc[x], ")"
)
})
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
plot.title = element_text(hjust = 0.5, colour = "black"),
legend.title = element_text(hjust = 0.5, colour = "black", angle = 0),
legend.text = element_text(colour = "black"),
legend.position = c(0.34, 0.16),
legend.background = element_blank(),
legend.key = element_rect(colour = "transparent", fill = "transparent")
) +
labs(fill = "") +
ylim(0, 1) +
xlab("Ranked known antigens (scaled)") +
ylab("Percent rank")p_combined <- plot_grid(p1, p2, nrow = 1, labels = c("a", "b"))
p_combinedpng(file = "./figures/Supplementary Fig 6.png", width = 7600, height = 3500, res = 600)
print(p_combined)
dev.off()
pdf(file = "../supplementary_figures/Supplementary Fig 6.pdf", width = 15.2, height = 7)
print(p_combined)
dev.off()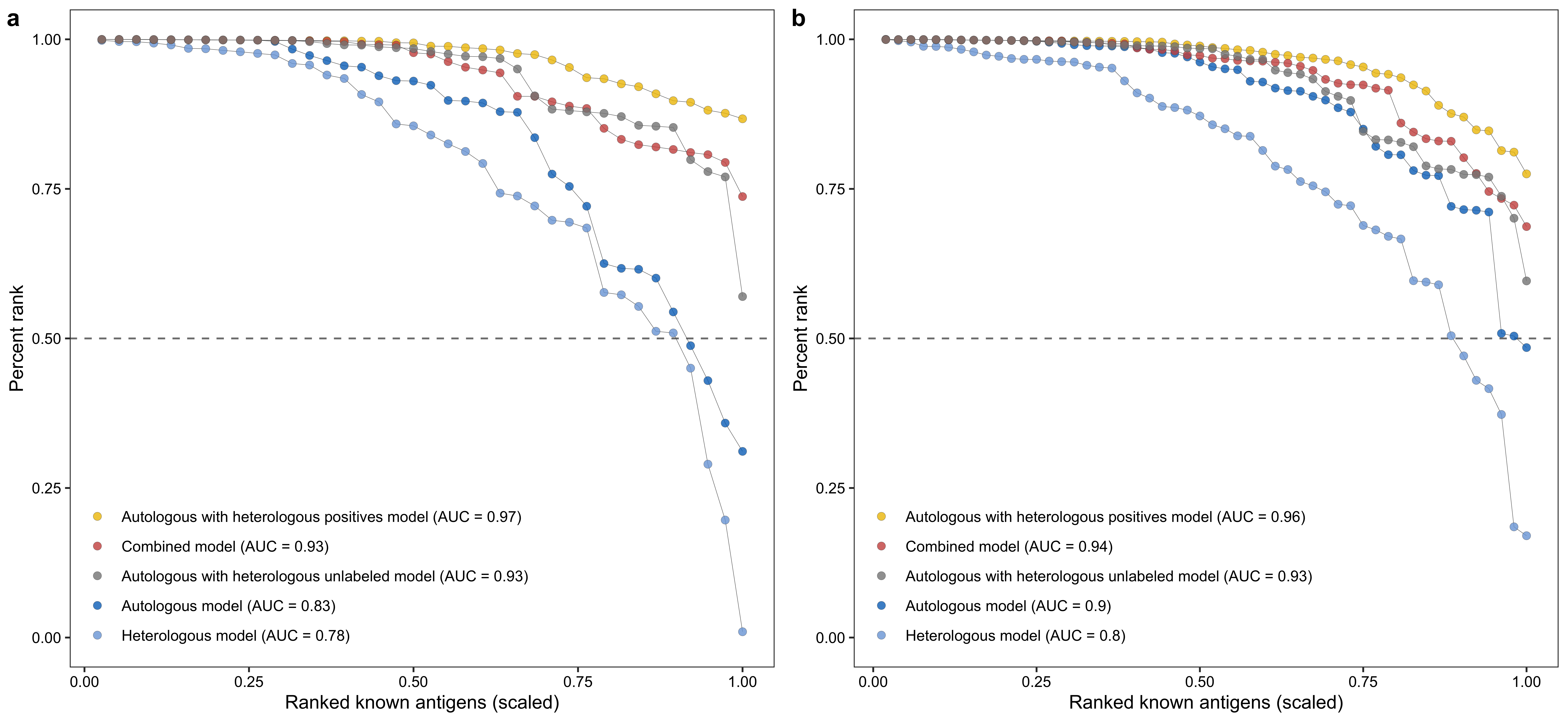
4.3 Score and species association
In R:
library(scales)
library(rlist)
library(rcompanion)
library(DT)scientific <- function(x) {
ifelse(x == 0, "0", gsub("e", " * 10^", scientific_format(digits = 3)(x)))
}
contingency_table <- list()
chisq_pval <- c()
cramer_res <- list()
# Combined
data <- read.csv("./data/supplementary_data_5_pfpv_purf_oob_predictions.csv", check.names = FALSE, row.names = 1)
pred <- sapply(data$`OOB score filtered`[data$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species <- data$species[data$antigen_label == 0]
M <- table(pred, species)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# P. vivax & P. falciparum single models
data_1 <- read.csv("./other_data/pf_single_pv_single_scores.csv", check.names = FALSE, row.names = 1)
data_2 <- read.csv("./other_data/pf_single_pv_single_cross_predictions.csv", check.names = FALSE, row.names = 1)
pred_1 <- sapply(data_1$`OOB score filtered`[data_1$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species_1 <- data_1$species[data_1$antigen_label == 0]
pred_2 <- sapply(data_2$`OOB score filtered`[data_2$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species_2 <- data_2$species[data_2$antigen_label == 0]
## P. vivax
M <- table(
c(pred_1[species_1 == "pv"], pred_2[species_2 == "pf"]),
c(rep("pv", length(pred_1[species_1 == "pv"])), rep("pf", length(pred_2[species_2 == "pf"])))
)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
## P. falciparum
M <- table(
c(pred_1[species_1 == "pf"], pred_2[species_2 == "pv"]),
c(rep("pf", length(pred_1[species_1 == "pf"])), rep("pv", length(pred_2[species_2 == "pv"])))
)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# P. vivax with heterologous positives
data <- read.csv("./other_data/pv_pf_pos_scores.csv", check.names = FALSE, row.names = 1)
pred <- sapply(data$`OOB scores`[data$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species <- data$species[data$antigen_label == 0]
M <- table(pred, species)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# P. falciparum with heterologous positives
data <- read.csv("./other_data/pf_pv_pos_scores.csv", check.names = FALSE, row.names = 1)
pred <- sapply(data$`OOB scores`[data$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species <- data$species[data$antigen_label == 0]
M <- table(pred, species)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# P. vivax with heterologous unlabeled
data <- read.csv("./other_data/pv_pf_unl_scores.csv", check.names = FALSE, row.names = 1)
pred <- sapply(data$`OOB scores`[data$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species <- data$species[data$antigen_label == 0]
M <- table(pred, species)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# P. falciparum with heterologous unlabeled
data <- read.csv("./other_data/pf_pv_unl_scores.csv", check.names = FALSE, row.names = 1)
pred <- sapply(data$`OOB scores`[data$antigen_label == 0], function(x) if (x >= 0.5) "pos" else "neg")
species <- data$species[data$antigen_label == 0]
M <- table(pred, species)
Xsq <- chisq.test(M, correct = FALSE)
cramerV <- cramerV(M, ci = TRUE)
contingency_table <- list.append(contingency_table, M)
chisq_pval <- c(chisq_pval, Xsq$p.value)
cramer_res <- list.append(cramer_res, sprintf("%0.2f", cramerV))
# Save results
df <- as.data.frame(cbind(
c(
"Combined", "P. vivax", "P. falciparum",
"P. vivax with heterologous positives",
"P. falciparum with heterologous positives",
"P. vivax with heterologous unlabeled",
"P. falciparum with heterologous unlabeled"
),
chisq_pval, do.call(rbind, cramer_res)
))
colnames(df) <- c("PURF model", "Chi-squared test p-value", "Cramer's V", "Lower CI", "Upper CI")
df$`Chi-squared test p-value` <- sapply(as.numeric(df$`Chi-squared test p-value`), scientific)
names(contingency_table) <- df$`PURF model`
save(contingency_table, df, file = "./rdata/score_species_association.RData")load("./rdata/score_species_association.RData")
contingency_table## $Combined
## species
## pred pf pv
## neg 2719 3814
## pos 2622 2639
##
## $`P. falciparum`
##
## pf pv
## neg 2897 3091
## pos 2444 3362
##
## $`P. vivax`
##
## pf pv
## neg 2451 3618
## pos 2890 2835
##
## $`P. falciparum with heterologous positives`
## species
## pred pf pv
## neg 2932 63
## pos 2409 6390
##
## $`P. vivax with heterologous positives`
## species
## pred pf pv
## neg 0 3695
## pos 5341 2758
##
## $`P. falciparum with heterologous unlabeled`
## species
## pred pf pv
## neg 108 6314
## pos 5233 139
##
## $`P. vivax with heterologous unlabeled`
## species
## pred pf pv
## neg 5133 1055
## pos 208 5398df %>%
datatable(rownames = FALSE)4.4 Tree depth analysis
4.4.1 Analysis
In Python:
library(reticulate)
use_condaenv("/Users/renee/Library/r-miniconda/envs/purf/bin/python")import pandas as pd
import numpy as np
import pickle
from purf.pu_ensemble import PURandomForestClassifier
import session_infowith open('./pickle_data/pf_0.5_purf_tree_filtering.pkl', 'rb') as infile:
pf_single_model = pickle.load(infile)
with open('./pickle_data/pv_0.5_purf_tree_filtering.pkl', 'rb') as infile:
pv_single_model = pickle.load(infile)
with open('./pickle_data/pfpv_0.5_purf_tree_filtering.pkl', 'rb') as infile:
pfpv_combined_model = pickle.load(infile)
with open('./pickle_data/pf_pv_pos_purf.pkl', 'rb') as infile:
pf_pv_pos_model = pickle.load(infile)
with open('./pickle_data/pf_pv_unl_purf.pkl', 'rb') as infile:
pf_pv_unl_model = pickle.load(infile)
with open('./pickle_data/pv_pf_pos_purf.pkl', 'rb') as infile:
pv_pf_pos_model = pickle.load(infile)
with open('./pickle_data/pv_pf_unl_purf.pkl', 'rb') as infile:
pv_pf_unl_model = pickle.load(infile)
res = pd.DataFrame({'pf_single_model': [tree.get_depth() for tree in pf_single_model['model'].estimators_],
'pv_single_model': [tree.get_depth() for tree in pv_single_model['model'].estimators_],
'pfpv_combined_model': [tree.get_depth() for tree in pfpv_combined_model['model'].estimators_],
'pf_pv_pos_model': [tree.get_depth() for tree in pf_pv_pos_model['model'].estimators_],
'pf_pv_unl_model': [tree.get_depth() for tree in pf_pv_unl_model['model'].estimators_],
'pv_pf_pos_model': [tree.get_depth() for tree in pv_pf_pos_model['model'].estimators_],
'pv_pf_unl_model': [tree.get_depth() for tree in pv_pf_unl_model['model'].estimators_]})
res.to_csv('./other_data/tree_depths.csv', index=False)In R
4.4.2 Plotting
library(psych)
library(ggrepel)data <- read.csv("./other_data/tree_depths.csv")
proportion_positives <- c(52 / 5393, 90 / 5431, 52 / 11846, 38 / 6491, 90 / 6543, 38 / 11832, 90 / 11884)
mean_tree_depth <- c(
mean(data$pf_single_model), mean(data$pf_pv_pos_model), mean(data$pf_pv_unl_model),
mean(data$pv_single_model), mean(data$pv_pf_pos_model), mean(data$pv_pf_unl_model),
mean(data$pfpv_combined_model)
)
sd_tree_depth <- c(
sd(data$pf_single_model), sd(data$pf_pv_pos_model), sd(data$pf_pv_unl_model),
sd(data$pv_single_model), sd(data$pv_pf_pos_model), sd(data$pv_pf_unl_model),
sd(data$pfpv_combined_model)
)
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("No transformation\n")
## No transformation
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = proportion_positives,
mean_tree_depth = mean_tree_depth
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = proportion_positives,
## mean_tree_depth = mean_tree_depth))
##
## Residuals:
## 1 2 3 4 5 6 7
## -0.12948 -0.97473 -1.25954 0.10225 0.04435 -1.13048 3.34764
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 3.269 1.411 2.317 0.0683 .
## proportion_positives 366.882 143.355 2.559 0.0507 .
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 1.735 on 5 degrees of freedom
## Multiple R-squared: 0.5671, Adjusted R-squared: 0.4805
## F-statistic: 6.55 on 1 and 5 DF, p-value: 0.05069
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("Log transform for tree depth\n")
## Log transform for tree depth
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = proportion_positives,
mean_tree_depth = log2(mean_tree_depth)
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = proportion_positives,
## mean_tree_depth = log2(mean_tree_depth)))
##
## Residuals:
## 1 2 3 4 5 6 7
## 0.058792 -0.267279 -0.329568 0.140857 -0.004416 -0.344751 0.746365
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 1.772 0.341 5.197 0.00348 **
## proportion_positives 94.203 34.650 2.719 0.04184 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.4194 on 5 degrees of freedom
## Multiple R-squared: 0.5965, Adjusted R-squared: 0.5158
## F-statistic: 7.391 on 1 and 5 DF, p-value: 0.04184
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("Logit transform for proportion positives\n")
## Logit transform for proportion positives
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = logit(proportion_positives),
mean_tree_depth = mean_tree_depth
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = logit(proportion_positives),
## mean_tree_depth = mean_tree_depth))
##
## Residuals:
## 1 2 3 4 5 6 7
## -0.6182 -0.7549 -1.0271 -0.0954 -0.1369 -0.2818 2.9143
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 22.7794 4.8653 4.682 0.00542 **
## proportion_positives 3.3428 0.9906 3.375 0.01979 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 1.457 on 5 degrees of freedom
## Multiple R-squared: 0.6949, Adjusted R-squared: 0.6339
## F-statistic: 11.39 on 1 and 5 DF, p-value: 0.01979
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("Arc-sine transform for proportion positives\n")
## Arc-sine transform for proportion positives
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = asin(sqrt(proportion_positives)),
mean_tree_depth = mean_tree_depth
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = asin(sqrt(proportion_positives)),
## mean_tree_depth = mean_tree_depth))
##
## Residuals:
## 1 2 3 4 5 6 7
## -0.37586 -0.90400 -1.11266 0.04187 -0.08305 -0.72069 3.15439
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -0.06706 2.29355 -0.029 0.9778
## proportion_positives 72.39644 24.52407 2.952 0.0318 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 1.592 on 5 degrees of freedom
## Multiple R-squared: 0.6354, Adjusted R-squared: 0.5625
## F-statistic: 8.715 on 1 and 5 DF, p-value: 0.03181
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("Logit transform for proportion positives & log transform for tree depth \n")
## Logit transform for proportion positives & log transform for tree depth
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = logit(proportion_positives),
mean_tree_depth = log2(mean_tree_depth)
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = logit(proportion_positives),
## mean_tree_depth = log2(mean_tree_depth)))
##
## Residuals:
## 1 2 3 4 5 6 7
## -0.06899 -0.21823 -0.26484 0.09247 -0.05659 -0.11887 0.63505
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 6.8270 1.1096 6.153 0.00165 **
## proportion_positives 0.8676 0.2259 3.840 0.01212 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.3322 on 5 degrees of freedom
## Multiple R-squared: 0.7468, Adjusted R-squared: 0.6962
## F-statistic: 14.75 on 1 and 5 DF, p-value: 0.01212
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
cat("Arc-sine transform for proportion positives & log transform for tree depth\n")
## Arc-sine transform for proportion positives & log transform for tree depth
cat("~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~\n")
## ~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~*~
res <- lm(mean_tree_depth ~ proportion_positives,
data = data.frame(
proportion_positives = asin(sqrt(proportion_positives)),
mean_tree_depth = log2(mean_tree_depth)
)
)
summary(res)
##
## Call:
## lm(formula = mean_tree_depth ~ proportion_positives, data = data.frame(proportion_positives = asin(sqrt(proportion_positives)),
## mean_tree_depth = log2(mean_tree_depth)))
##
## Residuals:
## 1 2 3 4 5 6 7
## -0.00527 -0.25295 -0.28949 0.12670 -0.03982 -0.23622 0.69705
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 0.9066 0.5417 1.673 0.1551
## proportion_positives 18.6878 5.7926 3.226 0.0233 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.3761 on 5 degrees of freedom
## Multiple R-squared: 0.6755, Adjusted R-squared: 0.6106
## F-statistic: 10.41 on 1 and 5 DF, p-value: 0.02331data <- data.frame(
proportion_positives = logit(proportion_positives),
mean_tree_depth = log2(mean_tree_depth),
upper_error_tree_depth = log2(mean_tree_depth + sd_tree_depth),
lower_error_tree_depth = log2(mean_tree_depth - sd_tree_depth)
)
data$model <- c(
"italic(P.~falciparum)~model",
"italic(P.~falciparum)~with~heterologous~positives~model",
"italic(P.~falciparum)~with~heterologous~unlabeled~model",
"italic(P.~vivax)~model",
"italic(P.~vivax)~with~heterologous~positives~model",
"italic(P.~vivax)~with~heterologous~unlabeled~model",
"Combined~model"
)
lm_eqn <- function(data) {
m <- lm(mean_tree_depth ~ proportion_positives, data)
eq <- substitute(
log[2](italic(y)) == a + b %.% logit(italic(x)) * "," ~ ~ italic(r^2) ~ "=" ~ r2,
list(
a = format(unname(coef(m)[1]), digits = 2),
b = format(unname(coef(m)[2]), digits = 2),
r2 = format(summary(m)$adj.r.squared, digits = 2)
)
)
as.character(as.expression(eq))
}
p <- ggplot(data, aes(proportion_positives, mean_tree_depth)) +
geom_smooth(method = "lm", formula = y ~ x, se = FALSE, color = "grey50", linetype = "dashed", linewidth = 0.5) +
geom_point(color = "grey10", size = 1.5, alpha = 0.8) +
geom_errorbar(aes(ymin = lower_error_tree_depth, ymax = upper_error_tree_depth), width = 0.05, linewidth = 0.2) +
geom_text_repel(aes(label = model),
size = 2.5, parse = TRUE, segment.size = 0.2,
nudge_x = 0.02, nudge_y = -0.15, point.padding = 0.1
) +
geom_text(x = -5.5, y = 3.5, label = lm_eqn(data), parse = TRUE, size = 3) +
scale_x_continuous(
breaks = logit(c(0.004, 0.008, 0.012, 0.016)),
labels = c(0.004, 0.008, 0.012, 0.016)
) +
scale_y_continuous(breaks = log2(c(0.25, 0.5, 2, 4, 8)), labels = c(0.25, 0.5, 2, 4, 8)) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.title = element_text(hjust = 0.5, colour = "black"),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black")
) +
xlab("Proportion of positives") +
ylab("Mean tree depth")png(file = "./figures/Supplementary Fig 7.png", width = 3800, height = 3500, res = 600)
print(p)
dev.off()
pdf(file = "../supplementary_figures/Supplementary Fig 7.pdf", width = 7.6, height = 7)
print(p)
dev.off()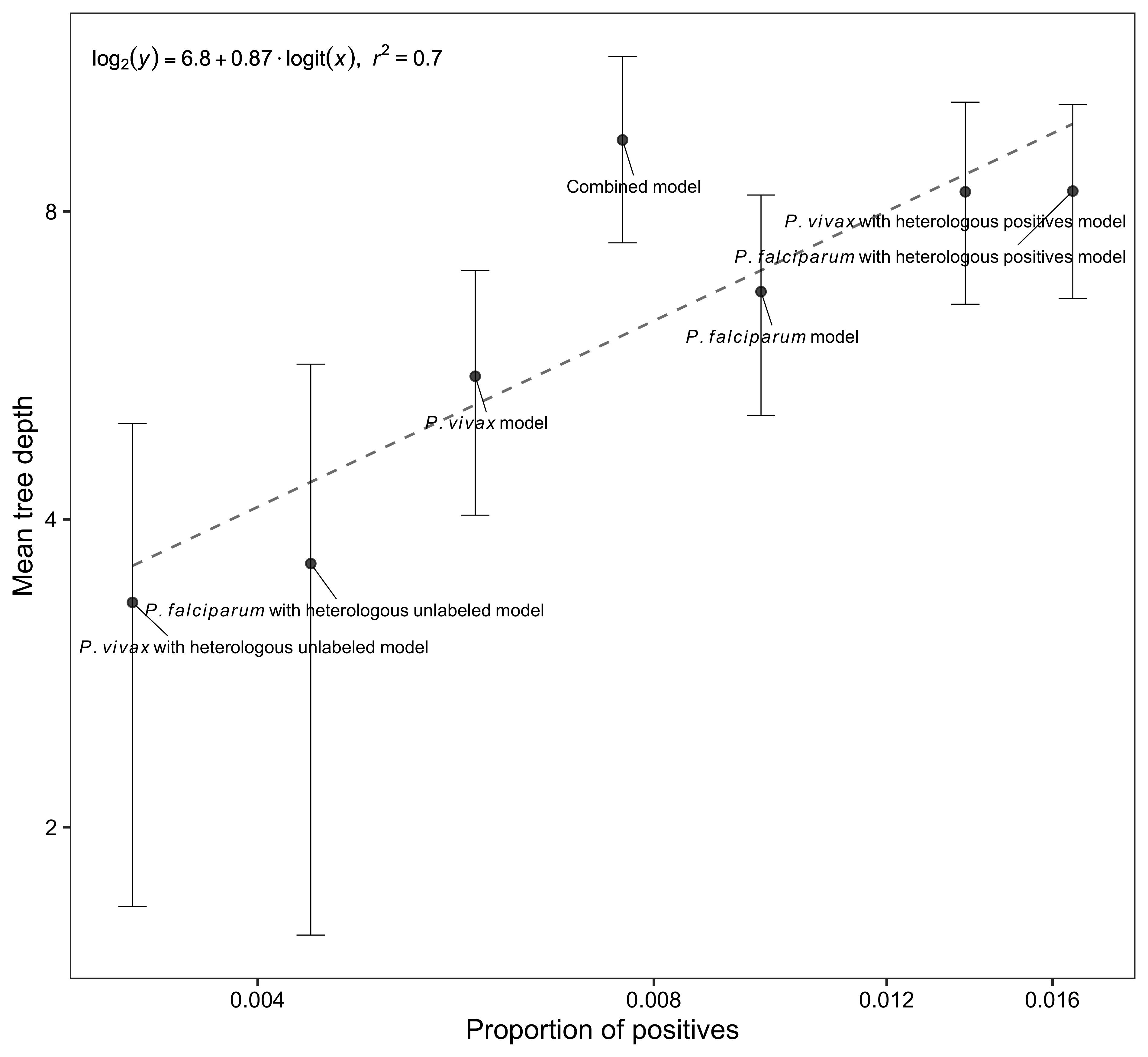
sessionInfo()## R version 4.2.3 (2023-03-15)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggrepel_0.9.3 psych_2.3.3 reticulate_1.28 DT_0.27
## [5] rcompanion_2.4.30 rlist_0.4.6.2 scales_1.2.1 ggsci_3.0.0
## [9] pracma_2.4.2 cowplot_1.1.1 gghalves_0.1.4 ggdist_3.2.1
## [13] reshape2_1.4.4 ggplot2_3.4.2
##
## loaded via a namespace (and not attached):
## [1] nlme_3.1-162 matrixStats_0.63.0 httr_1.4.6
## [4] rprojroot_2.0.3 R.cache_0.16.0 tools_4.2.3
## [7] bslib_0.4.2 utf8_1.2.3 R6_2.5.1
## [10] nortest_1.0-4 colorspace_2.1-0 withr_2.5.0
## [13] mnormt_2.1.1 tidyselect_1.2.0 Exact_3.2
## [16] compiler_4.2.3 cli_3.6.1 expm_0.999-7
## [19] sandwich_3.0-2 bookdown_0.34 sass_0.4.6
## [22] lmtest_0.9-40 mvtnorm_1.1-3 proxy_0.4-27
## [25] multcompView_0.1-9 stringr_1.5.0 digest_0.6.31
## [28] rmarkdown_2.21 R.utils_2.12.2 pkgconfig_2.0.3
## [31] htmltools_0.5.5 styler_1.9.1 fastmap_1.1.1
## [34] highr_0.10 htmlwidgets_1.6.2 rlang_1.1.1
## [37] readxl_1.4.2 rstudioapi_0.14 jquerylib_0.1.4
## [40] farver_2.1.1 generics_0.1.3 zoo_1.8-12
## [43] jsonlite_1.8.4 crosstalk_1.2.0 dplyr_1.1.2
## [46] R.oo_1.25.0 distributional_0.3.2 magrittr_2.0.3
## [49] modeltools_0.2-23 Matrix_1.5-4 Rcpp_1.0.10
## [52] DescTools_0.99.48 munsell_0.5.0 fansi_1.0.4
## [55] lifecycle_1.0.3 R.methodsS3_1.8.2 stringi_1.7.12
## [58] multcomp_1.4-23 yaml_2.3.7 MASS_7.3-60
## [61] rootSolve_1.8.2.3 plyr_1.8.8 grid_4.2.3
## [64] parallel_4.2.3 lmom_2.9 lattice_0.21-8
## [67] splines_4.2.3 knitr_1.42 pillar_1.9.0
## [70] boot_1.3-28.1 gld_2.6.6 codetools_0.2-19
## [73] stats4_4.2.3 glue_1.6.2 evaluate_0.21
## [76] data.table_1.14.8 vctrs_0.6.2 png_0.1-8
## [79] cellranger_1.1.0 gtable_0.3.3 purrr_1.0.1
## [82] cachem_1.0.8 xfun_0.39 coin_1.4-2
## [85] libcoin_1.0-9 e1071_1.7-13 class_7.3-22
## [88] survival_3.5-5 tibble_3.2.1 TH.data_1.1-2
## [91] ellipsis_0.3.2 here_1.0.1session_info.show()## -----
## numpy 1.19.0
## pandas 1.3.2
## purf NA
## session_info 1.0.0
## -----
## Python 3.8.2 (default, Mar 26 2020, 10:45:18) [Clang 4.0.1 (tags/RELEASE_401/final)]
## macOS-10.16-x86_64-i386-64bit
## -----
## Session information updated at 2023-05-18 12:35