Section 6 Top candidate comparisons
6.1 Clustering analysis
6.1.1 Analysis
In R:
library(DT)
library(factoextra)
library(NbClust)
library(cluster)
library(ggplot2)
library(cowplot)pfpv_data <- read.csv("./data/supplementary_data_5_pfpv_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)
pfpv_data <- pfpv_data[order(-pfpv_data$`OOB score filtered`), ]
pfpv_threshold <- pfpv_data[pfpv_data$antigen_label == 1, ]$`OOB score filtered`[sum(pfpv_data$antigen_label == 1) / 2]
top_pf_pfpv <- rownames(pfpv_data[pfpv_data$species == "pf" & pfpv_data$antigen_label == 0 & pfpv_data$`OOB score filtered` >= pfpv_threshold, ])
top_pv_pfpv <- rownames(pfpv_data[pfpv_data$species == "pv" & pfpv_data$antigen_label == 0 & pfpv_data$`OOB score filtered` >= pfpv_threshold, ])pfpv_prox <- read.csv("~/Downloads/pfpv_proximity_values.csv", check.names = FALSE)
pfpv_dist <- 1 - pfpv_prox
rownames(pfpv_dist) <- colnames(pfpv_dist)
pfpv_dist <- pfpv_dist[
rownames(pfpv_dist) %in% c(top_pf_pfpv, top_pv_pfpv),
colnames(pfpv_dist) %in% c(top_pf_pfpv, top_pv_pfpv)
]
rm(pfpv_prox)# Gap statistic
gap_stat <- clusGap(pfpv_dist, K.max = 10, hcut, B = 100, hc_method = "ward.D2")
p0_1 <- fviz_gap_stat(gap_stat, maxSE = list(method = "Tibs2001SEmax", SE.factor = 2)) +
labs(title = "Gap statistic method")
# Silhouette method
p0_2 <- fviz_nbclust(pfpv_dist, k.max = 10, hcut, hc_method = "ward.D2", method = "silhouette") +
labs(title = "Silhouette method")
# Elbow method
p0_3 <- fviz_nbclust(pfpv_dist, k.max = 10, hcut, hc_method = "ward.D2", method = "wss") +
geom_vline(xintercept = 3, linetype = 2, color = "steelblue") +
labs(title = "Elbow method")
save(p0_1, p0_2, p0_3, file = "./rdata/top_candidate_optimal_num_clusters.RData")library(umap)
library(ggplot2)
library(ggrepel)
library(cowplot)top_pfpv_hc <- hclust(as.dist(pfpv_dist), method = "ward.D2")
top_pfpv_2_clusters <- cutree(top_pfpv_hc, k = 2)
top_pfpv_3_clusters <- cutree(top_pfpv_hc, k = 3)
top_pfpv_2_clusters[top_pfpv_2_clusters == 1] <- 4
top_pfpv_2_clusters[top_pfpv_2_clusters == 2] <- 3
top_pfpv_2_clusters <- top_pfpv_2_clusters - 2
top_pfpv_3_clusters[top_pfpv_3_clusters == 1] <- 5
top_pfpv_3_clusters[top_pfpv_3_clusters == 2] <- 6
top_pfpv_3_clusters[top_pfpv_3_clusters == 3] <- 4
top_pfpv_3_clusters <- top_pfpv_3_clusters - 3
save(top_pfpv_2_clusters, top_pfpv_3_clusters,
file = "./rdata/top_candidate_clusters.RData"
)clusters <- top_pfpv_2_clusters
data <- read.csv("./data/supplementary_data_5_pfpv_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)
load("./rdata/pfpv_prox_mds_umap.RData")
umap_df_ <- umap_df
umap_df$label <- data$antigen_label
umap_df <- rbind(umap_df[umap_df$label == 1, ], umap_df[rownames(umap_df) %in% names(clusters), ])
umap_df$group <- ""
umap_df$group[umap_df$label == 1] <- "Known antigen"
umap_df$group[rownames(umap_df) %in% c("PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1", "PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1")] <- "Reference antigen"
umap_df$group[rownames(umap_df) %in% names(clusters)[clusters == 1]] <- "Group 1"
umap_df$group[rownames(umap_df) %in% names(clusters)[clusters == 2]] <- "Group 2"
umap_df_text <- umap_df[umap_df$label == 1, ]
umap_df_text$label_text <- ""
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0304600.1-p1"] <- "PfCSP"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0424100.1-p1"] <- "RH5"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0206900.1-p1"] <- "MSP5"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0209000.1-p1"] <- "P230"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0835600.1-p1"] <- "PvCSP"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0623800.1-p1"] <- "DBP"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0728900.1-p1"] <- "MSP1"
umap_p1 <- ggplot() +
geom_point(data = umap_df_, aes(x = X1, y = X2), color = "grey90", alpha = 0.5) +
geom_point(
data = umap_df[umap_df$label == 1, ], aes(x = X1, y = X2), shape = 21, color = "black",
fill = "#FFFF33", stroke = 0.3, size = 2, alpha = 0.5
) +
geom_point(data = umap_df[umap_df$label == 0, ], aes(x = X1, y = X2, fill = group), shape = 21, alpha = 0.5) +
geom_text_repel(
data = umap_df_text, aes(x = X1, y = X2, label = label_text), size = 3.5,
nudge_x = 1.5, nudge_y = -1, point.padding = 0.1
) +
scale_fill_manual(breaks = c("Group 1", "Group 2"), values = c("#03a1fc", "#984EA3")) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(title = "")) +
xlab("Dimension 1") +
ylab("Dimension 2")clusters <- top_pfpv_3_clusters
data <- read.csv("./data/supplementary_data_5_pfpv_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)
load("./rdata/pfpv_prox_mds_umap.RData")
umap_df_ <- umap_df
umap_df$label <- data$antigen_label
umap_df <- rbind(umap_df[umap_df$label == 1, ], umap_df[rownames(umap_df) %in% names(clusters), ])
umap_df$group <- ""
umap_df$group[umap_df$label == 1] <- "Known antigen"
umap_df$group[rownames(umap_df) %in% c("PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1", "PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1")] <- "Reference antigen"
umap_df$group[rownames(umap_df) %in% names(clusters)[clusters == 1]] <- "Group 1"
umap_df$group[rownames(umap_df) %in% names(clusters)[clusters == 2]] <- "Group 2"
umap_df$group[rownames(umap_df) %in% names(clusters)[clusters == 3]] <- "Group 3"
umap_df_text <- umap_df[umap_df$label == 1, ]
umap_df_text$label_text <- ""
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0304600.1-p1"] <- "PfCSP"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0424100.1-p1"] <- "RH5"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0206900.1-p1"] <- "MSP5"
umap_df_text$label_text[rownames(umap_df_text) == "PF3D7_0209000.1-p1"] <- "P230"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0835600.1-p1"] <- "PvCSP"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0623800.1-p1"] <- "DBP"
umap_df_text$label_text[rownames(umap_df_text) == "PVP01_0728900.1-p1"] <- "MSP1"
umap_p2 <- ggplot() +
geom_point(data = umap_df_, aes(x = X1, y = X2), color = "grey90", alpha = 0.5) +
geom_point(
data = umap_df[umap_df$label == 1, ], aes(x = X1, y = X2), shape = 21, color = "black",
fill = "#FFFF33", stroke = 0.3, size = 2, alpha = 0.5
) +
geom_point(data = umap_df[umap_df$label == 0, ], aes(x = X1, y = X2, fill = group), shape = 21, alpha = 0.5) +
geom_text_repel(
data = umap_df_text, aes(x = X1, y = X2, label = label_text), size = 3.5,
nudge_x = 1.5, nudge_y = -1, point.padding = 0.1
) +
scale_fill_manual(
breaks = c("Group 1", "Group 2", "Group 3"),
values = c("#03a1fc", "#984EA3", "#FF7F00")
) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
plot.margin = ggplot2::margin(5, 5, 5, 5, "pt"),
axis.title = element_text(colour = "black"),
axis.text = element_text(colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(title = "")) +
xlab("Dimension 1") +
ylab("Dimension 2")umap_p_combined <- plot_grid(umap_p1, umap_p2, nrow = 1, labels = c("a", "b"))
umap_p_combined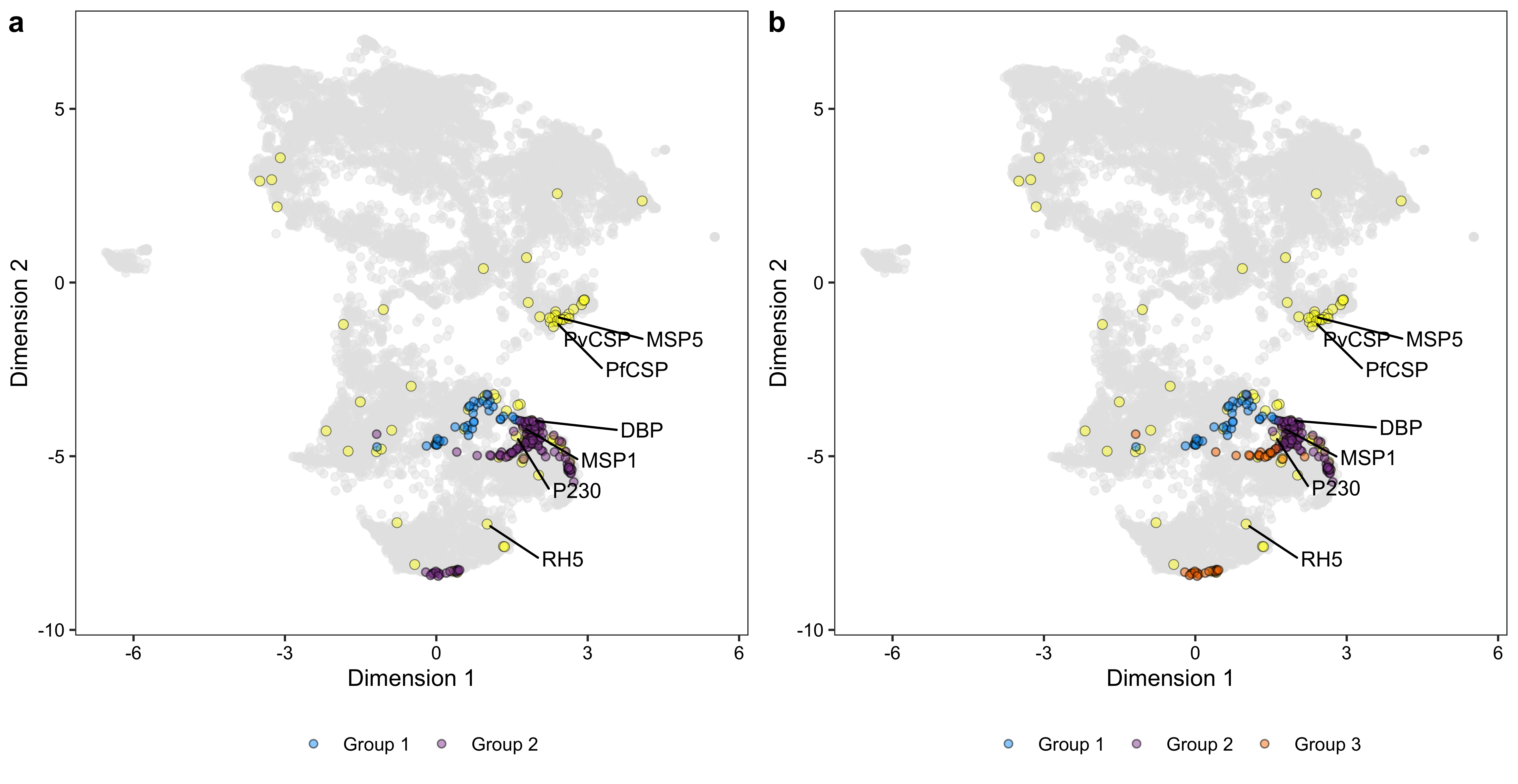
load(file = "./rdata/top_candidate_clusters.RData")
species <- c(rep("pf", 145), rep("pv", 45))
cat("Two clusters\n")## Two clusterstable(top_pfpv_2_clusters, species)## species
## top_pfpv_2_clusters pf pv
## 1 0 35
## 2 145 10cat("Three clusters\n")## Three clusterstable(top_pfpv_3_clusters, species)## species
## top_pfpv_3_clusters pf pv
## 1 0 35
## 2 107 9
## 3 38 1protein_id_1 <- names(top_pfpv_3_clusters)[top_pfpv_3_clusters == 1]
protein_id_2 <- names(top_pfpv_3_clusters)[top_pfpv_3_clusters == 2]
protein_id_3 <- names(top_pfpv_3_clusters)[top_pfpv_3_clusters == 3]
save(protein_id_1, protein_id_2, protein_id_3, file = "./rdata/clustering_groups.RData")
gene_accession_1 <- str_replace_all(protein_id_1, "\\.[1-9]-p1", "")
gene_accession_2 <- str_replace_all(protein_id_2, "\\.[1-9]-p1", "")
gene_accession_3 <- str_replace_all(protein_id_3, "\\.[1-9]-p1", "")
write.table(data.frame("acc" = gene_accession_1),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_candidates_gene_accession_1.csv"
)
write.table(data.frame("acc" = gene_accession_2),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_candidates_gene_accession_2.csv"
)
write.table(data.frame("acc" = gene_accession_3),
sep = ", ", row.names = FALSE, col.names = FALSE,
file = "./other_data/top_candidates_gene_accession_3.csv"
)6.2 Gene ontology analysis
6.2.1 Analysis
In Bash:
python ./other_data/goea.py -s "./other_data/top_candidates_gene_accession_1.csv" \
-p "./other_data/PlasmoDB-62_pfpv_GO.gaf" \
-n "./other_data/go-basic.obo" \
-o "./other_data/goea_result_1.xlsx"
python ./other_data/goea.py -s "./other_data/top_candidates_gene_accession_2.csv" \
-p "./other_data/PlasmoDB-62_pfpv_GO.gaf" \
-n "./other_data/go-basic.obo" \
-o "./other_data/goea_result_2.xlsx"
python ./other_data/goea.py -s "./other_data/top_candidates_gene_accession_3.csv" \
-p "./other_data/PlasmoDB-62_pfpv_GO.gaf" \
-n "./other_data/go-basic.obo" \
-o "./other_data/goea_result_3.xlsx"6.2.2 Plotting
In R:
library(ggplot2)
library(gridExtra)
library(shadowtext)
library(ggforce)
library(reshape)
library(cowplot)
library(scales)process_goea_res <- function(file_name) {
data <- read.csv(file_name, header = TRUE, fill = TRUE, quote = '\"', stringsAsFactors = FALSE)
# calculate percentage from the ratio column
data$num_genes <- apply(data, 1, function(x) as.integer(unlist(strsplit(x["ratio_in_study"], "/"))[1]))
# select for enrichment (e) data rows
data <- data[data$enrichment == "e", ]
# select for biological process (BP) data rows
res_bp <- data[data$NS == "BP", ]
res_bp$`-Log10FDR` <- -log10(res_bp$p_fdr_bh)
res_bp <- res_bp[order(res_bp$`-Log10FDR`), ]
res_bp$group <- rep("Biological process", nrow(res_bp))
# select for cellular component (CC) data rows
res_cc <- data[data$NS == "CC", ]
res_cc$`-Log10FDR` <- -log10(res_cc$p_fdr_bh)
res_cc <- res_cc[order(res_cc$`-Log10FDR`), ]
res_cc$group <- rep("Cellular component", nrow(res_cc))
# select for molecular function (MF) data rows
res_mf <- data[data$NS == "MF", ]
res_mf$`-Log10FDR` <- -log10(res_mf$p_fdr_bh)
res_mf <- res_mf[order(res_mf$`-Log10FDR`), ]
res_mf$group <- rep("Molecular function", nrow(res_mf))
ds <- do.call(rbind, list(res_mf, res_cc, res_bp))
ds$name <- factor(ds$name, levels = ds$name)
ds$group <- factor(ds$group, levels = c("Biological process", "Cellular component", "Molecular function"))
return(ds)
}ds_1 <- process_goea_res("./other_data/goea_result_1.csv")
ds_2 <- process_goea_res("./other_data/goea_result_2.csv")
ds_3 <- process_goea_res("./other_data/goea_result_3.csv")p1 <- ggplot(ds_1, aes(x = name, y = `-Log10FDR`)) +
geom_col(width = 0.05) +
geom_point(size = 5, shape = 21, color = "black", fill = "grey20") +
geom_text(aes(label = sprintf("%.0f", num_genes)), nudge_y = 0, size = 2.5, color = "white") +
scale_x_discrete(labels = label_wrap(50)) +
coord_flip() +
ggforce::facet_col(facets = vars(group), scales = "free_y", space = "free") +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.spacing = unit(0, "pt"),
strip.background = element_blank(),
panel.border = element_rect(colour = "black", size = 0.5),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(5, 5, 85, 5, "pt"),
legend.text = element_text(colour = "black", ),
axis.title.x = element_text(colour = "black", ),
axis.title.y = element_text(colour = "black", ),
axis.text.x = element_text(colour = "black", ),
axis.text.y = element_text(colour = "black", )
) +
xlab("") +
ylab(expression("-Log"[10] * "FDR")) +
ggtitle("Group 1 (35 candidates)")p2 <- ggplot(ds_2, aes(x = name, y = `-Log10FDR`)) +
geom_col(width = 0.05) +
geom_point(size = 5, shape = 21, color = "black", fill = "grey20") +
geom_text(aes(label = sprintf("%.0f", num_genes)), nudge_y = 0, size = 2.5, color = "white") +
scale_x_discrete(labels = label_wrap(50)) +
coord_flip() +
ggforce::facet_col(facets = vars(group), scales = "free_y", space = "free") +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.spacing = unit(0, "pt"),
strip.background = element_blank(),
panel.border = element_rect(colour = "black", size = 0.5),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(5, 0, 5, 5, "pt"),
legend.text = element_text(colour = "black", ),
axis.title.x = element_text(colour = "black", ),
axis.title.y = element_text(colour = "black", ),
axis.text.x = element_text(colour = "black", ),
axis.text.y = element_text(colour = "black", )
) +
xlab("") +
ylab(expression("-Log"[10] * "FDR")) +
ggtitle("Group 2 (116 candidates)")p3 <- ggplot(ds_3, aes(x = name, y = `-Log10FDR`)) +
geom_col(width = 0.05) +
geom_point(size = 5, shape = 21, color = "black", fill = "grey20") +
geom_text(aes(label = sprintf("%.0f", num_genes)), nudge_y = 0, size = 2.5, color = "white") +
scale_x_discrete(labels = label_wrap(50)) +
coord_flip() +
ggforce::facet_col(facets = vars(group), scales = "free_y", space = "free") +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.spacing = unit(0, "pt"),
strip.background = element_blank(),
panel.border = element_rect(colour = "black", size = 0.5),
plot.title = element_text(hjust = 0.5),
plot.margin = ggplot2::margin(5, 5, 5, 0, "pt"),
legend.text = element_text(colour = "black", ),
axis.title.x = element_text(colour = "black", ),
axis.title.y = element_text(colour = "black", ),
axis.text.x = element_text(colour = "black", ),
axis.text.y = element_text(colour = "black", )
) +
xlab("") +
ylab(expression("-Log"[10] * "FDR")) +
ggtitle("Group 3 (39 candidates)")Final plot
p_combined <- plot_grid(p1, p2, p3, nrow = 1, rel_widths = c(0.3, 0.3, 0.3), labels = c("a", "b", "c"))
p_combined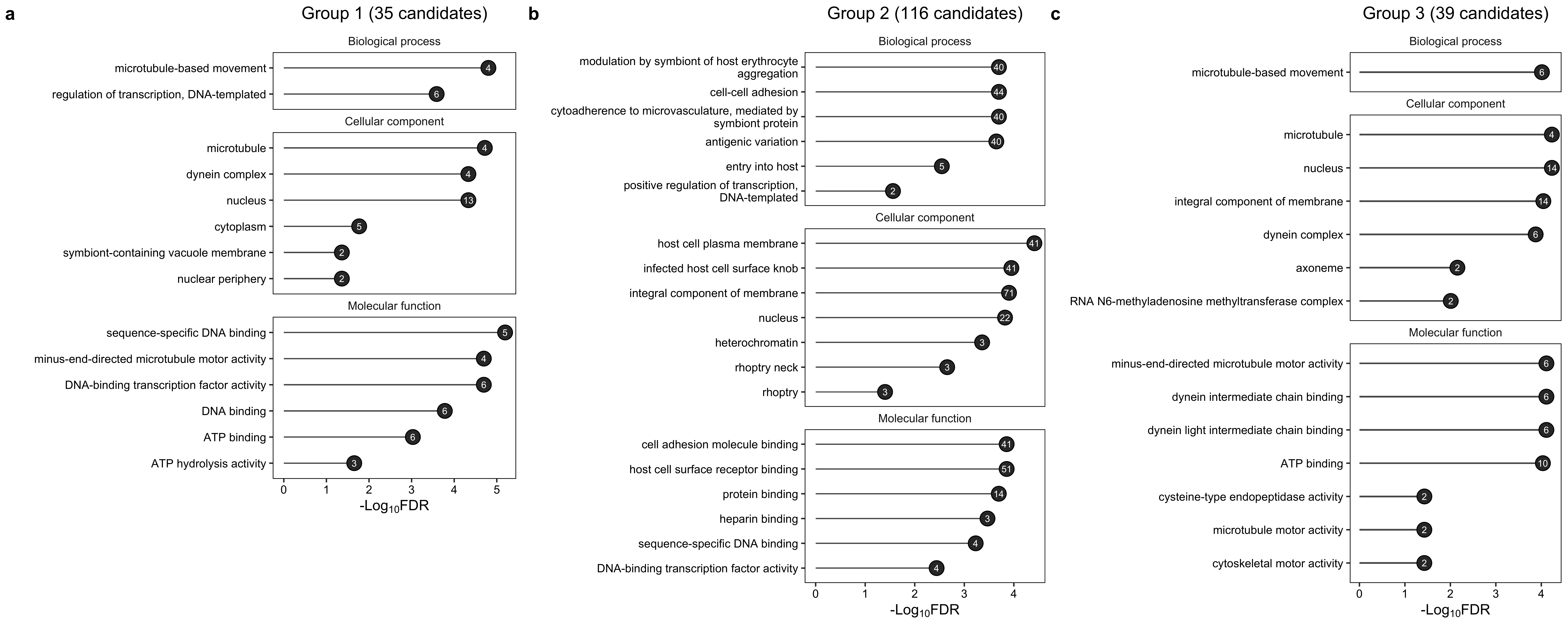
6.3 Candidate antigen characterization
6.3.1 Processing table
In R:
library(tibble)prediction <- read.csv("./data/supplementary_data_5_pfpv_purf_oob_predictions.csv", row.names = 1, check.names = FALSE)
prediction <- prediction[order(-prediction$`OOB score filtered`), ]
threshold <- prediction[prediction$antigen_label == 1, ]$`OOB score filtered`[sum(prediction$antigen_label == 1) / 2]
labeled_pos <- rownames(prediction)[prediction$antigen_label == 1]
top_pfpv <- rownames(prediction[prediction$antigen_label == 0 & prediction$`OOB score filtered` >= threshold, ])pfpv_prox <- read.csv("~/Downloads/pfpv_proximity_values.csv", check.names = FALSE)
pfpv_dist <- 1 - pfpv_prox
rm(pfpv_prox)
rownames(pfpv_dist) <- colnames(pfpv_dist)
pfpv_dist <- pfpv_dist[, labeled_pos]ds <- prediction[c(labeled_pos, top_pfpv), "OOB score filtered", drop = FALSE]
ds <- rownames_to_column(ds, "Protein ID")
colnames(ds)[2] <- "Score"
# Add clustering groups
load(file = "./rdata/clustering_groups.RData")
ds$Group <- ""
ds$Group[ds$`Protein ID` %in% protein_id_1] <- "Group 1"
ds$Group[ds$`Protein ID` %in% protein_id_2] <- "Group 2"
ds$Group[ds$`Protein ID` %in% protein_id_3] <- "Group 3"
ds$Group[ds$`Protein ID` %in% labeled_pos] <- "Known antigen"
ds$Group[ds$`Protein ID` %in% c(
"PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1",
"PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1",
"PVP01_0835600.1-p1", "PVP01_0623800.1-p1",
"PVP01_0728900.1-p1"
)] <- "Reference antigen"
# Add gene products
gene_products <- rbind(
read.csv("./other_data/pf3d7_gene_products_v62.csv"),
read.csv("./other_data/pvp01_gene_products_v62.csv")
)
colnames(gene_products) <- c("Protein ID", "Gene product")
ds <- merge(x = ds, y = gene_products, by = "Protein ID", all.x = TRUE)
# Add closest reference antigen and the distance
pfpv_ad_ctrl_res <- read.csv("./other_data/pfpv_ad_ctrl_res.csv", check.names = FALSE, row.names = 1)
ds$`Closest known antigen` <- ""
ds$`Known antigen source` <- "Intersect"
ds$`Closest distance` <- -1
for (i in 1:nrow(ds)) {
prot <- ds$`Protein ID`[i]
tmp <- pfpv_dist[prot, ]
ds[i, "Closest known antigen"] <- names(tmp)[which.min(tmp)]
ds[i, "Closest known antigen"] <- paste0(
ds[i, "Closest known antigen"], " (",
gene_products[
gene_products$`Protein ID` == ds[i, "Closest known antigen"],
"Gene product"
], ")"
)
if (names(tmp)[which.min(tmp)] %in% c(
"PF3D7_0304600.1-p1", "PF3D7_0424100.1-p1",
"PF3D7_0206900.1-p1", "PF3D7_0209000.1-p1",
"PVP01_0835600.1-p1", "PVP01_0623800.1-p1",
"PVP01_0728900.1-p1"
)) {
ds[i, "Known antigen source"] <- "Reference"
} else {
ds[i, "Known antigen source"] <- pfpv_ad_ctrl_res[names(tmp)[which.min(tmp)], "source"]
}
ds[i, "Closest distance"] <- tmp[which.min(tmp)]
}
# Sort based on scores
ds <- ds[order(-ds$Score), ]
write.csv(ds, file = "./data/supplementary_data_6_antigen_characterization.csv", row.names = FALSE)sessionInfo()## R version 4.2.3 (2023-03-15)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] tibble_3.2.1 scales_1.2.1 reshape_0.8.9 ggforce_0.4.1
## [5] shadowtext_0.1.2 gridExtra_2.3 ggrepel_0.9.3 umap_0.2.10.0
## [9] cowplot_1.1.1 cluster_2.1.4 NbClust_3.0.1 factoextra_1.0.7
## [13] ggplot2_3.4.2 DT_0.27
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.10 lattice_0.21-8 png_0.1-8 digest_0.6.31
## [5] utf8_1.2.3 RSpectra_0.16-1 R6_2.5.1 plyr_1.8.8
## [9] evaluate_0.21 highr_0.10 pillar_1.9.0 rlang_1.1.1
## [13] rstudioapi_0.14 jquerylib_0.1.4 R.utils_2.12.2 R.oo_1.25.0
## [17] Matrix_1.5-4 reticulate_1.28 rmarkdown_2.21 styler_1.9.1
## [21] htmlwidgets_1.6.2 polyclip_1.10-4 munsell_0.5.0 compiler_4.2.3
## [25] xfun_0.39 pkgconfig_2.0.3 askpass_1.1 htmltools_0.5.5
## [29] openssl_2.0.6 tidyselect_1.2.0 bookdown_0.34 codetools_0.2-19
## [33] fansi_1.0.4 dplyr_1.1.2 withr_2.5.0 MASS_7.3-60
## [37] R.methodsS3_1.8.2 grid_4.2.3 jsonlite_1.8.4 gtable_0.3.3
## [41] lifecycle_1.0.3 magrittr_2.0.3 cli_3.6.1 cachem_1.0.8
## [45] farver_2.1.1 bslib_0.4.2 generics_0.1.3 vctrs_0.6.2
## [49] tools_4.2.3 R.cache_0.16.0 glue_1.6.2 tweenr_2.0.2
## [53] purrr_1.0.1 fastmap_1.1.1 yaml_2.3.7 colorspace_2.1-0
## [57] knitr_1.42 sass_0.4.6